5. Single-molecule data analysis 2#
[1]:
from pathlib2 import Path
import papylio as pp
import matplotlib.pyplot as plt
import numpy as np
import xarray as xr
%matplotlib inline
Experiment import#
Note that the sequencing data is automatically imported.
[2]:
experiment_path = Path(r'C:\Users\user\Desktop\SPARXS example dataset')
[3]:
data_per_sequence_path = experiment_path / 'Analysis' / 'Datasets per sequence'
data_per_sequence_path
[3]:
WindowsPath('C:/Users/user/Desktop/SPARXS example dataset/Analysis/Datasets per sequence')
[4]:
exp = pp.Experiment(data_per_sequence_path)
Import files: 100%|█████████████████████████████████████████████████████████████| 7948/7948 [00:00<00:00, 46126.39it/s]
Initialize experiment:
C:\Users\user\Desktop\SPARXS example dataset\Analysis\Datasets per sequence
[5]:
exp.files[0:10]
[5]:
FileCollection([File(AAAAAGCG),
File(AAAAAGTT),
File(AAACAAAA),
File(AAAGCATC),
File(AAAGGAGT),
File(AAATCGCT),
File(AAATCTAT),
File(AAATTCGG),
File(AAATTGCG),
File(AACAACGT)])
[6]:
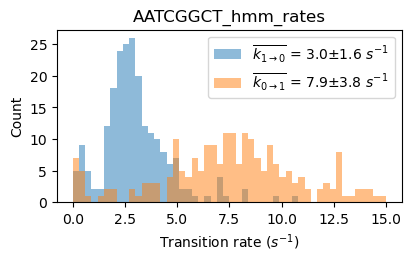
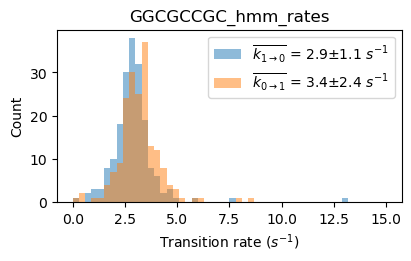
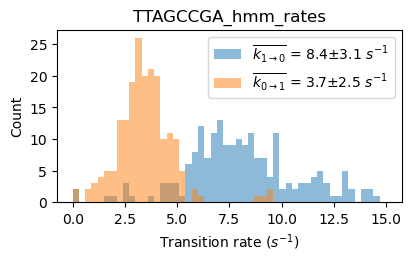
file_HJ1 = exp.files.select('TTAGCCGA', 'name')[0]
file_HJ3 = exp.files.select('AATCGGCT', 'name')[0]
file_HJ7 = exp.files.select('GGCGCCGC', 'name')[0]
files_HJ137 = exp.files.select('TTAGCCGA|AATCGGCT|GGCGCCGC', 'name')
Molecule selection (or filtering)#
Determine intensity thresholds#
The thresholds are generally best determined from the data originating from all sequences, as opposed to some example sequences.
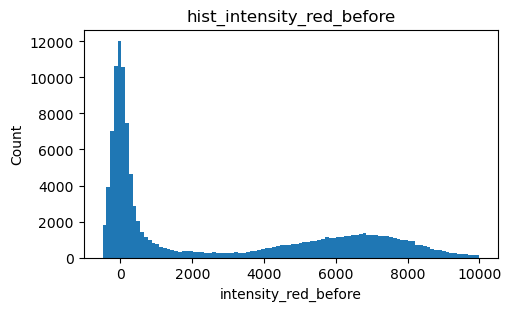
For the Holliday junction dataset we selected molecules for which the Cy5 acceptor was active both before and after imaging with the green laser. In this way we did not have to take acceptor bleaching into account. Additionally, we selected molecules with total intensity (Cy3 + Cy5) upon green laser excitation, to be in the single-molecule range, i.e. above background and without other molecules present. Hence, we needed to determine three thresholds, a lower treshold for Cy5 and an lower and upper threshold for the total intensity.
Cy5 intensity before imaging with green laser
[7]:
intensity_red_before = exp.files.intensity_red_before.sel(channel=1)
100%|██████████████████████████████████████████████████████████████████████████████| 7948/7948 [01:19<00:00, 99.92it/s]
[8]:
fig, ax = plt.subplots(figsize=(5, 3), layout='constrained')
intensity_red_before.plot.hist(bins=100, ax=ax, range=(-500,10000))
ax.set_ylabel('Count')
title = 'hist_intensity_red_before'
ax.set_title(title)
[8]:
Text(0.5, 1.0, 'hist_intensity_red_before')
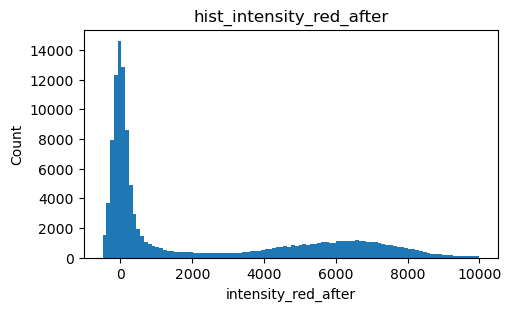
Cy5 intensity after imaging with the green laser
[9]:
intensity_red_after = exp.files.intensity_red_after.sel(channel=1)
100%|█████████████████████████████████████████████████████████████████████████████| 7948/7948 [01:10<00:00, 112.68it/s]
[10]:
fig, ax = plt.subplots(figsize=(5, 3), layout='constrained')
intensity_red_after.plot.hist(bins=100, ax=ax, range=(-500,10000))
ax.set_ylabel('Count')
title = 'hist_intensity_red_after'
ax.set_title(title)
[10]:
Text(0.5, 1.0, 'hist_intensity_red_after')
Total intensity
[11]:
intensity = exp.files.intensity
100%|█████████████████████████████████████████████████████████████████████████████| 7948/7948 [01:13<00:00, 108.72it/s]
[12]:
intensity_total_start = intensity.sel(frame=np.arange(5,10)).sum('channel').mean('frame')
[13]:
fig, ax = plt.subplots(figsize=(5, 3), layout='constrained')
c, x, _ = intensity_total_start.plot.hist(bins=100, ax=ax, range=(0,50000))# ,histtype='step')
x_center = (x[1:] + x[:-1])/2
def gaussian(x, A, mu, sig):
return A * np.exp(-(x-mu)**2 / (2 * sig **2))
selection = (x_center>11000) & (x_center<20000)
from scipy.optimize import curve_fit
popt, pcov = curve_fit(gaussian, x_center[selection], c[selection], p0=(np.max(c), np.mean(x_center), np.mean(x_center)))
ax.plot(x_center, gaussian(x_center, *popt))
ax.set_xlabel('Intensity total')
ax.set_ylabel('Count')
A, mu, sig = popt
sig = np.abs(sig)
print('Intensity range 3 sigma', mu - 3 * sig, mu + 3 * sig)
# title = 'Intensity range 3 sigma'
# fig.savefig(save_path / (title + '.png'))
# fig.savefig(save_path / (title + '.pdf'))
Intensity range 3 sigma 5665.467330207128 29523.580994683183
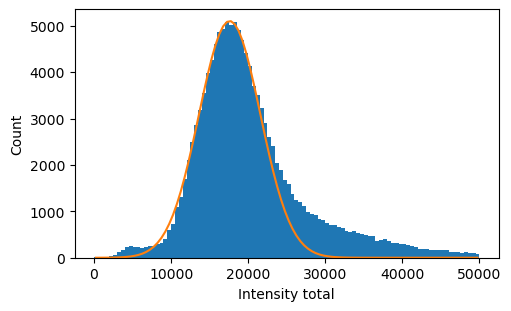
Finally, we selected the following tresholds.
[14]:
red_intensity_cutoff = 2000
intensity_total_min = 9000
intensity_total_max = 30000
Define selections#
As an example we perform this on HJ7 file.
[15]:
file = file_HJ7
[16]:
selection_Cy5_active_start = file.intensity_red_before.sel(channel=1, drop=True) > red_intensity_cutoff
selection_Cy5_active_start.name = 'selection_Cy5_active_start'
selection_Cy5_active_start
[16]:
<xarray.DataArray 'selection_Cy5_active_start' (molecule: 753)>
array([False, False, True, True, True, False, False, False, False,
False, False, False, True, True, False, True, False, False,
True, False, False, False, True, True, False, False, False,
False, False, True, True, True, False, False, False, False,
True, False, True, True, True, False, True, False, True,
True, True, True, True, False, False, False, False, True,
False, False, False, True, True, False, True, True, True,
True, True, True, False, True, False, True, False, True,
True, True, True, False, True, False, True, False, True,
False, True, True, False, True, False, False, False, False,
True, False, False, False, False, True, True, False, True,
False, True, True, True, False, True, True, True, True,
True, False, True, True, True, False, True, True, False,
False, False, False, True, True, False, False, True, True,
True, False, True, True, False, True, True, True, True,
True, False, True, True, True, False, False, True, True,
True, True, False, False, False, True, True, True, True,
True, False, False, False, False, True, True, False, False,
False, True, True, True, True, False, True, True, False,
False, False, False, False, False, True, True, True, False,
...
False, True, True, True, False, False, True, False, True,
False, False, True, False, False, True, False, True, True,
True, True, True, False, False, False, False, True, True,
True, False, False, False, False, True, True, True, False,
False, True, True, True, True, True, False, False, False,
False, False, False, True, True, False, True, True, False,
True, True, False, False, True, True, False, False, False,
False, False, True, False, True, False, False, False, True,
False, True, True, True, False, True, True, True, True,
True, False, True, True, False, True, True, False, False,
True, False, False, False, False, False, True, False, True,
True, True, False, False, False, True, True, False, True,
True, True, True, True, False, True, False, False, True,
False, False, False, False, True, True, True, True, False,
False, True, True, True, False, False, False, True, False,
True, True, False, False, False, True, True, False, True,
True, True, True, False, False, True, True, True, False,
True, True, True, False, False, False, False, False, True,
False, True, False, False, True, True, True, True, False,
False, True, True, True, True, False])
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule- molecule: 753
- False False True True True False ... False True True True True False
array([False, False, True, True, True, False, False, False, False, False, False, False, True, True, False, True, False, False, True, False, False, False, True, True, False, False, False, False, False, True, True, True, False, False, False, False, True, False, True, True, True, False, True, False, True, True, True, True, True, False, False, False, False, True, False, False, False, True, True, False, True, True, True, True, True, True, False, True, False, True, False, True, True, True, True, False, True, False, True, False, True, False, True, True, False, True, False, False, False, False, True, False, False, False, False, True, True, False, True, False, True, True, True, False, True, True, True, True, True, False, True, True, True, False, True, True, False, False, False, False, True, True, False, False, True, True, True, False, True, True, False, True, True, True, True, True, False, True, True, True, False, False, True, True, True, True, False, False, False, True, True, True, True, True, False, False, False, False, True, True, False, False, False, True, True, True, True, False, True, True, False, False, False, False, False, False, True, True, True, False, ... False, True, True, True, False, False, True, False, True, False, False, True, False, False, True, False, True, True, True, True, True, False, False, False, False, True, True, True, False, False, False, False, True, True, True, False, False, True, True, True, True, True, False, False, False, False, False, False, True, True, False, True, True, False, True, True, False, False, True, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, True, True, False, True, True, False, True, True, False, False, True, False, False, False, False, False, True, False, True, True, True, False, False, False, True, True, False, True, True, True, True, True, False, True, False, False, True, False, False, False, False, True, True, True, True, False, False, True, True, True, False, False, False, True, False, True, True, False, False, False, True, True, False, True, True, True, True, False, False, True, True, True, False, True, True, True, False, False, False, False, False, True, False, True, False, False, True, True, True, True, False, False, True, True, True, True, False]) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
[17]:
selection_Cy5_active_end = file.intensity_red_after.sel(channel=1, drop=True) > red_intensity_cutoff
selection_Cy5_active_end.name = 'selection_Cy5_active_end'
selection_Cy5_active_end
[17]:
<xarray.DataArray 'selection_Cy5_active_end' (molecule: 753)>
array([False, False, True, True, True, False, False, False, False,
False, False, False, True, True, False, True, False, False,
True, False, False, False, True, False, False, False, False,
False, False, False, True, False, False, False, False, False,
True, False, True, True, True, False, True, False, True,
True, True, True, True, False, False, False, False, True,
False, False, False, True, True, False, False, True, False,
True, True, True, False, True, False, True, False, True,
True, True, True, False, True, False, True, False, True,
False, True, True, False, False, False, False, False, False,
True, False, False, False, False, True, True, False, True,
False, False, True, True, False, True, False, True, True,
False, False, True, True, True, False, True, True, False,
False, False, False, True, False, False, False, True, True,
True, False, True, False, False, True, True, True, True,
True, False, False, True, True, False, False, True, True,
True, True, False, False, False, True, True, True, True,
True, False, False, False, True, False, True, False, False,
False, True, True, True, True, False, True, True, False,
False, False, False, False, False, True, True, True, False,
...
False, True, True, True, False, False, False, False, True,
False, False, False, False, False, False, False, True, True,
True, True, False, False, False, False, False, True, True,
True, False, False, False, False, False, True, True, False,
False, True, True, False, True, True, False, False, False,
False, False, False, True, True, False, True, True, False,
True, True, False, False, True, True, False, False, False,
False, False, True, False, True, False, False, False, True,
False, True, True, True, False, True, True, True, False,
True, False, True, True, False, True, False, False, False,
True, False, False, False, False, False, False, False, False,
True, False, False, False, False, True, True, False, True,
True, True, False, True, False, False, False, False, True,
False, False, False, False, False, False, True, True, False,
False, True, True, True, False, False, False, False, False,
True, True, False, False, False, True, True, False, True,
False, True, True, False, False, True, True, True, False,
True, True, True, False, False, False, False, False, True,
False, True, False, False, True, True, True, True, False,
False, True, True, True, True, False])
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule- molecule: 753
- False False True True True False ... False True True True True False
array([False, False, True, True, True, False, False, False, False, False, False, False, True, True, False, True, False, False, True, False, False, False, True, False, False, False, False, False, False, False, True, False, False, False, False, False, True, False, True, True, True, False, True, False, True, True, True, True, True, False, False, False, False, True, False, False, False, True, True, False, False, True, False, True, True, True, False, True, False, True, False, True, True, True, True, False, True, False, True, False, True, False, True, True, False, False, False, False, False, False, True, False, False, False, False, True, True, False, True, False, False, True, True, False, True, False, True, True, False, False, True, True, True, False, True, True, False, False, False, False, True, False, False, False, True, True, True, False, True, False, False, True, True, True, True, True, False, False, True, True, False, False, True, True, True, True, False, False, False, True, True, True, True, True, False, False, False, True, False, True, False, False, False, True, True, True, True, False, True, True, False, False, False, False, False, False, True, True, True, False, ... False, True, True, True, False, False, False, False, True, False, False, False, False, False, False, False, True, True, True, True, False, False, False, False, False, True, True, True, False, False, False, False, False, True, True, False, False, True, True, False, True, True, False, False, False, False, False, False, True, True, False, True, True, False, True, True, False, False, True, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, False, True, False, True, True, False, True, False, False, False, True, False, False, False, False, False, False, False, False, True, False, False, False, False, True, True, False, True, True, True, False, True, False, False, False, False, True, False, False, False, False, False, False, True, True, False, False, True, True, True, False, False, False, False, False, True, True, False, False, False, True, True, False, True, False, True, True, False, False, True, True, True, False, True, True, True, False, False, False, False, False, True, False, True, False, False, True, True, True, True, False, False, True, True, True, True, False]) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
[18]:
intensity_total_rolling = file.intensity_total.rolling(frame=5, center=True).mean().dropna('frame')
selection_intensity_total = (intensity_total_rolling < intensity_total_max).all('frame')
selection_intensity_total.name = 'selection_intensity_total'
selection_intensity_total
[18]:
<xarray.DataArray 'selection_intensity_total' (molecule: 753)>
array([ True, True, True, False, True, True, True, True, False,
True, True, True, True, True, False, False, False, True,
True, True, True, True, True, True, True, False, False,
True, True, True, True, False, True, False, True, False,
True, True, True, False, False, True, True, False, True,
False, True, True, True, True, True, True, False, True,
True, True, True, True, True, True, True, True, False,
True, True, False, True, True, True, True, True, True,
True, True, True, True, True, True, False, True, True,
False, True, False, False, False, True, True, True, True,
True, True, True, True, True, True, True, True, False,
True, True, True, True, True, True, True, False, True,
True, True, True, True, True, True, True, False, True,
False, True, True, True, True, True, True, True, True,
True, True, True, True, True, True, True, True, False,
True, True, True, True, True, False, True, True, True,
True, False, False, True, True, True, True, True, True,
False, True, True, False, True, True, True, False, True,
True, True, True, True, True, True, True, False, True,
False, True, True, True, True, True, True, False, True,
...
True, True, True, True, True, True, True, True, True,
True, True, True, False, True, True, True, True, True,
True, False, False, False, True, True, True, True, True,
True, True, False, True, True, False, False, True, True,
True, True, True, False, True, True, False, True, False,
False, False, False, False, False, True, True, False, True,
True, True, True, True, False, True, True, True, True,
True, True, True, True, True, True, True, True, True,
True, True, True, True, False, True, True, True, True,
False, True, True, True, True, True, True, True, True,
False, True, False, False, True, True, True, True, False,
True, True, True, True, True, False, False, True, True,
True, False, True, False, True, True, True, True, True,
True, True, True, True, True, True, False, True, True,
True, True, True, False, False, False, True, False, True,
True, True, True, True, False, True, True, True, True,
True, True, True, True, True, False, True, True, True,
False, True, False, True, True, True, False, True, True,
False, False, True, False, True, True, True, True, False,
True, True, False, True, False, True])
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule- molecule: 753
- True True True False True True ... True True False True False True
array([ True, True, True, False, True, True, True, True, False, True, True, True, True, True, False, False, False, True, True, True, True, True, True, True, True, False, False, True, True, True, True, False, True, False, True, False, True, True, True, False, False, True, True, False, True, False, True, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, False, True, True, False, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, False, True, False, False, False, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, False, True, True, True, True, False, False, True, True, True, True, True, True, False, True, True, False, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, True, True, True, True, True, True, False, True, ... True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, True, False, False, False, True, True, True, True, True, True, True, False, True, True, False, False, True, True, True, True, True, False, True, True, False, True, False, False, False, False, False, False, True, True, False, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, False, True, True, True, True, False, True, True, True, True, True, False, False, True, True, True, False, True, False, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, False, False, False, True, False, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, False, True, True, True, False, True, False, True, True, True, False, True, True, False, False, True, False, True, True, True, True, False, True, True, False, True, False, True]) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
Save and apply selections#
[19]:
file.selected
[19]:
<xarray.DataArray 'selected' (molecule: 753)>
array([False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
...
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False, False, False, False,
False, False, False, False, False, False])
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule- molecule: 753
- False False False False False False ... False False False False False
array([False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, ... False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False]) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
[20]:
file.set_variable(selection_Cy5_active_start)
file.set_variable(selection_Cy5_active_end)
file.set_variable(selection_intensity_total)
The selection are now stored in the dataset for this sequence, starting with selection_:
[21]:
file.dataset
[21]:
<xarray.Dataset>
Dimensions: (molecule: 753, channel: 2, dimension: 2,
frame: 400)
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 ... 848 1340 352
file (molecule) |S58 b'Single-molecule data - gree...
* channel (channel) int64 0 1
* dimension (dimension) |S1 b'x' b'y'
time (frame) float64 0.0 0.123 0.245 ... 48.75 48.87
* frame (frame) int32 0 1 2 3 4 ... 395 396 397 398 399
illumination (frame) int32 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0
sequence_in_file (molecule) int32 386 412 949 970 ... 160 758 670
Dimensions without coordinates: molecule
Data variables: (12/19)
selected (molecule) bool False False ... False False
coordinates (molecule, channel, dimension) float64 24.92 ...
intensity (molecule, channel, frame) float64 1.559e+04 ...
intensity_raw (molecule, channel, frame) float64 9.597e+03 ...
FRET (molecule, frame) float64 0.0 0.0 ... 0.05922
intensity_red_before (molecule, channel) float64 5.718 ... -143.0
... ...
sequence_subset (molecule) |S8 b'GGCGCCGC' ... b'GGCGCCGC'
sequence_quality_subset (molecule) |S8 b'7EGG<B7>' ... b'6@8C7C@B'
sequence_coordinates (molecule, dimension) int64 6849 2670 ... 24880
selection_Cy5_active_start (molecule) bool False False True ... True False
selection_Cy5_active_end (molecule) bool False False True ... True False
selection_intensity_total (molecule) bool True True True ... False True- molecule: 753
- channel: 2
- dimension: 2
- frame: 400
- molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - channel(channel)int640 1
array([0, 1], dtype=int64)
- dimension(dimension)|S1b'x' b'y'
array([b'x', b'y'], dtype='|S1')
- time(frame)float640.0 0.123 0.245 ... 48.75 48.87
- units :
- s
array([ 0. , 0.123, 0.245, 0.368, 0.49 , 0.612, 0.735, 0.858, 0.98 , 1.102, 1.225, 1.347, 1.47 , 1.592, 1.715, 1.838, 1.96 , 2.082, 2.205, 2.327, 2.45 , 2.573, 2.694, 2.817, 2.94 , 3.062, 3.185, 3.307, 3.429, 3.552, 3.675, 3.797, 3.92 , 4.042, 4.165, 4.287, 4.41 , 4.532, 4.654, 4.777, 4.9 , 5.022, 5.145, 5.267, 5.389, 5.512, 5.635, 5.757, 5.879, 6.002, 6.124, 6.247, 6.37 , 6.491, 6.614, 6.737, 6.859, 6.982, 7.104, 7.226, 7.349, 7.472, 7.594, 7.717, 7.839, 7.961, 8.084, 8.207, 8.329, 8.451, 8.574, 8.696, 8.819, 8.942, 9.063, 9.186, 9.309, 9.431, 9.553, 9.676, 9.799, 9.921, 10.044, 10.166, 10.288, 10.411, 10.534, 10.656, 10.778, 10.901, 11.024, 11.146, 11.269, 11.391, 11.513, 11.636, 11.759, 11.881, 12.003, 12.126, 12.248, 12.371, 12.494, 12.615, 12.738, 12.861, 12.983, 13.106, 13.228, 13.35 , 13.473, 13.596, 13.718, 13.841, 13.963, 14.085, 14.208, 14.331, 14.453, 14.575, 14.698, 14.82 , 14.943, 15.066, 15.187, 15.31 , 15.433, 15.555, 15.678, 15.8 , 15.922, 16.045, 16.168, 16.291, 16.413, 16.535, 16.658, 16.78 , 16.903, 17.026, 17.147, 17.27 , 17.393, 17.515, 17.638, 17.76 , 17.882, 18.005, 18.128, 18.25 , 18.372, 18.495, 18.618, 18.74 , 18.862, 18.985, 19.107, 19.23 , 19.353, 19.474, ... 29.396, 29.518, 29.64 , 29.763, 29.886, 30.008, 30.131, 30.253, 30.375, 30.498, 30.621, 30.743, 30.865, 30.988, 31.11 , 31.233, 31.356, 31.477, 31.6 , 31.723, 31.846, 31.968, 32.09 , 32.213, 32.335, 32.458, 32.581, 32.703, 32.825, 32.948, 33.07 , 33.193, 33.316, 33.437, 33.56 , 33.683, 33.805, 33.928, 34.05 , 34.172, 34.295, 34.418, 34.54 , 34.662, 34.785, 34.907, 35.03 , 35.153, 35.274, 35.397, 35.52 , 35.643, 35.765, 35.887, 36.01 , 36.132, 36.255, 36.378, 36.499, 36.622, 36.745, 36.867, 36.99 , 37.112, 37.234, 37.357, 37.48 , 37.602, 37.725, 37.847, 37.969, 38.092, 38.215, 38.337, 38.459, 38.582, 38.704, 38.827, 38.95 , 39.071, 39.194, 39.317, 39.439, 39.562, 39.685, 39.806, 39.929, 40.052, 40.174, 40.297, 40.419, 40.541, 40.664, 40.787, 40.909, 41.031, 41.154, 41.276, 41.399, 41.522, 41.644, 41.766, 41.889, 42.012, 42.134, 42.256, 42.379, 42.501, 42.624, 42.747, 42.868, 42.991, 43.114, 43.236, 43.359, 43.482, 43.603, 43.726, 43.849, 43.971, 44.094, 44.216, 44.338, 44.461, 44.584, 44.706, 44.828, 44.951, 45.073, 45.196, 45.319, 45.44 , 45.563, 45.686, 45.809, 45.931, 46.053, 46.176, 46.298, 46.421, 46.544, 46.665, 46.788, 46.911, 47.033, 47.156, 47.278, 47.4 , 47.523, 47.646, 47.768, 47.89 , 48.013, 48.135, 48.258, 48.381, 48.502, 48.625, 48.748, 48.87 ]) - frame(frame)int320 1 2 3 4 5 ... 395 396 397 398 399
array([ 0, 1, 2, ..., 397, 398, 399])
- illumination(frame)int320 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0
array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]) - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
- selected(molecule)boolFalse False False ... False False
array([False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, ... False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False, False]) - coordinates(molecule, channel, dimension)float6424.92 341.5 283.8 ... 441.4 130.2
array([[[ 24.91602904, 341.48554967], [283.78574085, 339.137688 ]], [[ 78.17807134, 372.90395929], [336.78871819, 370.50780467]], [[ 92.01072464, 166.17974665], [350.74208901, 164.28798759]], ..., [[ 25.45838149, 469.78131449], [284.34290719, 467.1585397 ]], [[ 22.58199174, 129.18829072], [281.6534317 , 127.36065026]], [[183.17939012, 132.032136 ], [441.40299696, 130.15824268]]]) - intensity(molecule, channel, frame)float641.559e+04 1.252e+04 ... 884.1
- background_correction :
- [-300 -100]
- gamma_correction :
- 1.5
- alpha_correction :
- 0.05
array([[[ 1.55884625e+04, 1.25170638e+04, 1.25944564e+04, ..., 1.64734899e+02, 2.69480270e+02, -4.69745833e+02], [-1.14025092e+03, -1.79434893e+03, 7.10999508e+02, ..., -2.62304725e+03, -1.10312074e+03, -6.55675284e+02]], [[ 1.34390768e+04, 1.35416917e+04, 1.74872344e+04, ..., 1.56752700e+04, 1.63730120e+04, 1.66206681e+04], [-3.65498855e+01, -1.07613788e+03, 7.68370384e+02, ..., -1.29670147e+02, -6.38025128e+02, -2.18210334e+02]], [[ 1.15077644e+04, 9.18438669e+03, 9.73603353e+03, ..., 1.01456006e+04, 1.09333859e+04, 1.25990483e+04], [ 1.11070711e+04, 1.09403092e+04, 8.19838649e+03, ..., 6.23282590e+03, 1.21338662e+04, 1.14991815e+04]], ..., [[ 1.00809311e+04, 9.02672236e+03, 1.28988727e+04, ..., 1.20465479e+04, 1.01208656e+04, 9.69752167e+03], [ 4.66614740e+03, 7.48326565e+03, 5.04780621e+03, ..., 6.04748313e+03, 6.15144622e+03, 5.87020252e+03]], [[ 2.36006570e+04, 1.70832674e+04, 4.73061255e+04, ..., 1.34665923e+04, 1.30160141e+04, 1.55987137e+04], [ 1.41042367e+04, 1.63379632e+04, 7.00118411e+03, ..., 9.50686972e+03, 1.23659826e+04, 1.21960524e+04]], [[ 1.79517282e+04, 1.74761086e+04, 1.71619744e+04, ..., 1.65578237e+04, 1.52834714e+04, 1.40444913e+04], [ 1.54934599e+02, 5.60340529e+02, 2.93738407e+02, ..., 3.17724251e+01, -7.98358369e+02, 8.84103653e+02]]]) - intensity_raw(molecule, channel, frame)float649.597e+03 7.647e+03 ... 1.486e+03
array([[[ 9597.43649264, 7647.34209132, 7696.48025203, ..., -195.40641347, -128.90141607, -598.25132255], [ -460.82779502, -1268.49574232, 1240.72232826, ..., -2714.81050151, -1189.64672654, -779.16257571]], [[ 8232.74718725, 8297.89946185, 10803.00594487, ..., 9652.55236575, 10095.56319399, 10252.80514106], [ 535.40395552, -499.0533011 , 1542.73210166, ..., 554.09335198, 80.62547306, 512.82307128]], [[ 7006.51706311, 5531.35663118, 5881.60858793, ..., 6141.65119131, 6641.8323304 , 7699.39571645], [11582.45931601, 11299.52852699, 8585.18816608, ..., 6640.10592707, 12580.53554253, 12029.13387182]], ..., [[ 6100.59119767, 5431.2522946 , 7889.76041476, ..., 7348.60182896, 6125.94639233, 5857.15661519], [ 5070.19395799, 7834.60176685, 5592.74984693, ..., 6549.81052174, 6557.48949828, 6255.07860606]], [[14684.54410822, 10546.51897643, 29735.63522788, ..., 8250.21732206, 7964.13593817, 9603.94521165], [15184.26951661, 17092.12655598, 9266.49038866, ..., 10080.19933784, 12916.78333291, 12875.98811846]], [[11097.92266571, 10795.9419459 , 10596.49170729, ..., 10212.90391941, 9403.79135695, 8617.1373611 ], [ 952.52100907, 1334.14595752, 1051.83712911, ..., 759.6636088 , -134.1848 , 1486.32822049]]]) - FRET(molecule, frame)float640.0 0.0 0.05344 ... 0.0 0.05922
array([[0. , 0. , 0.05343669, ..., 1.06701138, 1.32325718, 0.58260439], [0. , 0. , 0.04208956, ..., 0. , 0. , 0. ], [0.49114092, 0.54362606, 0.4571314 , ..., 0.38055096, 0.52602131, 0.47717951], ..., [0.31641165, 0.45325688, 0.28126687, ..., 0.33422531, 0.37803149, 0.37707519], [0.37406913, 0.48884984, 0.1289179 , ..., 0.41381964, 0.48719503, 0.43878953], [0.00855677, 0.03106712, 0.01682764, ..., 0.0019152 , 0. , 0.05922216]]) - intensity_red_before(molecule, channel)float645.718 435.4 35.45 ... 76.4 -143.0
array([[ 5.71771351e+00, 4.35361883e+02], [ 3.54482215e+01, -1.41182975e+02], [-2.72161708e+01, 7.28438296e+03], ..., [ 6.17814544e+01, 4.85823584e+03], [ 1.73779796e+01, 6.37530474e+03], [ 7.64042010e+01, -1.43028955e+02]]) - intensity_red_after(molecule, channel)float6449.42 103.4 -14.57 ... 13.65 111.7
array([[ 49.42475415, 103.44932566], [ -14.57350384, 244.16745202], [ 21.27267505, 6472.26269457], ..., [ 52.97241636, 4773.01440448], [ 60.07183329, 5599.77061963], [ 13.64937453, 111.68762698]]) - sequence_name(molecule)|S10b'HJ_general' ... b'HJ_general'
array([b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', ... b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general'], dtype='|S10') - sequence_tile(molecule)int641101 1101 1101 ... 1102 1102 1102
array([1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, ... 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102], dtype=int64) - sequence(molecule)|S147b'TACCATCCCAGGCATGTCCACCCACCGCTC...
array([b'TACCATCCCAGGCATGTCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGGGCGAGCGGGGGGAGATAGGAAGAGCAGATTCTGAACTAC', b'TACCATGCTCGCCAGCTCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCCA', b'TACCAATCAAGTGATGTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTAAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCACACCTGGGACATCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACT', b'TACCAGGCGAGGAATGTATACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCATACCTGCAAGTTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCACCCCAGTTACCTAGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAAATCGGAAGAGCACACGTCTGAACTC', b'TACCAGACCTGTGAAGTCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCAGCCTTGGCACGTCCACCCACCGCTCGGCTCAACTGGGTTTGTTGAGCGCTTGCTAGGGTTTTGCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGGGGGAGATCGGAAGAGCACACGTCTGAACTCCAGTCA', b'TACCAGGCCGGTCATATTAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAAACTGGCTAATTGTACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAACCCGGATACCTAGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAGACAGGGCACCTTGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCAATCCGGTGAGTTAAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCACCCGTGTGATTTTGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCACACGCGCTAAGTCGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAGCCAAGACATCTCAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCCA', b'TGTTTAGTAACGTACGCGCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAAACGAACTCGTATGCCGTCCTCTGCT', b'TACCACTCCAGCAATTTGCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAAGCAGGTAAAATATACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', ... b'TACCAACCGAGGTAATTCTACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGGGCGGTGGGAGATCGGAAGAGCAGCCGTCTGAACTCC', b'TACCATCCACGCCATCTCCACCCACCTCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAAATC', b'TACCAACCACGGGAGTTTTACCCACCGCTCGGCTCAACTGGGTTTTACCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCATGCTCGTGAGCTTAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCCGCGCGAGCGGTGGGAGATCGGAAGAGAACACGTCTTAACTC', b'TACAATCACGTGAGTTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCA', b'TACCATCCACGCGAATTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGACGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAAATC', b'TACCAAACTTGTCAACTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCCGTAGCAGCGCGGGCGGTGGGAAATCGGAAGAGCACACTTCTGAACTC', b'TACCAGACCGGATATCTTCACCCACCGCTCGGCTCAACTGGGTTTTCCCAATTGCGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCACTCTTGCGAACTTCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGCGCACACGTCTGAACTCC', b'TACCAGTCCCGCGATCTTGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGTTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAGACAGGACATGTTTACCCACCGCTCGGCTCAACTGGGTTTTCCCTGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTCGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCATTCGGGACACTTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCAAGCACGGTAGGTTAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCATTCTGGGGATATCAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTCTCCCTAGCAAGCCGCTGCTCCGGTTTTCCGTAGCCGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTT', b'TACCATACGTGTGACTTACACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGAAGCGCGAGCGGGGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACAAGCCCAGTAATTTAGACCCACCGCTCGGCTCAACTGGGTTTTCCTAGTTGAGCGCTTGCTAGGATTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCCCATGTCAGCTAAACCCACCGCTCGGCTCAACTGGGTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTATGAACTACAG', b'TACCAACCCTGCCATGTGTACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGATAAGGTTCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGCGATCGGAAGAGCACACGTT', b'TACCATGCCAGCTAGGTGGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCATAGCAAGCCGATGCTACGGTTTTCCGTAGCGGCGCGAGCGGTTGGGAATCGGGTGAGCAAACGTCTGAACTT'], dtype='|S147') - sequence_quality(molecule)|S147b'AB@<6-C,6CFGCGGGGCGGGGGFGCF+@F...
array([b'AB@<6-C,6CFGCGGGGCGGGGGFGCF+@F7EGGGDFFGFGGCBFGGGFGGFGFGGGGGGGDGGGGGG<FGGGGGGGCEGGG<BF@CGGGGGGEFCECFGGGGCD<A+7>>+@44F+3@8@78:,=C:+*42=<*++,75B<*00<<', b'CCCCCGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGFCFGGGG=EGFGGGGGGGGGFGGGGFGFGGGGGGGGGGGGFGGGGGGGGGGGGFEGGGGGGFEAAE@>EG7C=6:=F@ECDFFFDE:=DFFFDGG@@FD=E=<<E3', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGFEGGGGFGGGGGFGGGGGGGGFGGGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGECFFGGGGGGFGGGGGDFFGG@FGGCEGG98>=B@ECEGGGGGFGGFGGF83', b'CCCCCGGGGGEFFGGGGGGGGGGGGGGGGGECDCFFFGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCFGFFCFCGBC,9EFEGGGFFFGGDC@ECDEC74==CFFDEBDE7,2@C7@FFGD8EGDC8+8?FFG7', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGGGGFGGFEFFGGGGGGGGGGGEGGDFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEEEGGCFGGFCCEGDEGEGGGGGGGGFBF8:FGEF9', b'CCCCCGGGGGGGGGGFFGGGGGGGGGGEGGGGGGGGGGGGGGFBECFGGGGAFGGEFGGEGGGGGGGGGFGGGGGFGGGGGGCEFGGGGGGGGGGGGGFGGGGG7BEFE>@CEFGGCFGGGGG8ED>CCEFGGGGGGC=DF8DFD,3', b'CCCCCGGGGGGGFAFFGGGGGGGGGGGGFGC@FGGGGFGGGGFG7<FGEGGFFGGEAFGEEGGCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGEEBFC>:CEGGFEGGFGFGF@CFBCFFGGGGFGFFFFGG8==', b'@CCCCDGDGGFFFFGGFGGFGGGGGGGGGG@CFGGGGGGGGGFGGGGGGFGFFGGFFFGGGEFCFGGGGGGGGGGGGGGGGGEFGGGGGGGGGGGGGGGGGDEGGGFCFGGEGGGFEEFGGE,CF@CFEGDD@=DFGGGGFFGD@EE', b'CCCCCFDFFGFGEGFDGG@FGGGGGGGGGGC@EGGGGGGGGGFCGD=?FFF,CC@CDGAEEF?=B:FGAF@FGGFG7DEGCCEFFGGF,@7,<:FFCGGAFFGG4=F:CG7>F<:FE+2BFDEGG76FGFCE@**;AFC00<>+<C0', b'CCCCCGGGEFGGGGGGGGGGGGGGGDGGGGEDGGGGGGCCFFFEGG9F@EEEEFGGGDECFGG9<FFGCFGFF<FGGGGGGGFEEGFFGGGGEEFGGGFGGGGGGF,=>==:=F:F@=FG?CEFE@=FE>DGFGCGGFDFC4CFGC,', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCGEGGGGGGGGGGGDCGGGDCE8CFGGGGGGCFGGFDGEEA', b'CCCCCGGGGGDGEGGGGGGGGGGGGGGGFGCGFFGFGGGGGGFGGG9FFGG<FGGGGGGGGGGGGGGGEFG?F@BEFGGGGDCGGEEGDGGGGGGGGGGGGGC8DFFFEC>=EGGGGGGE88EFFDCGEGGFFGGFFFFCF4+@@E;', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGEEGGGDGGGGGGGG@FGGGGGGF<FFGGGGG>GFFGCGGFFFGGGGGGGGAFGGGGGDGGGGGGGGGGGGGGFGGGGFFDGGGGGGG=CEGGGG,CFCGGDFF,@8DFG8D8DEF?DFE', b'CCCCCGFFGGGGGGGFGGGGGGGGGGGGGGGEGGGGGGGGGGFFGCGGGGGGFGGGGGGGGFGGGGGGEFGFGGGGGGGGGGGGGGGGGGGGGG8CGGGGEFGGGGFFCECCEFGGCFGE:>=,C6@FEEGGGGGF?F=EFFDGAA,', b'CCCCCGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGFGFEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFEGGGGGGGGD=FGGGGGGGGGGGGGFGGGGGGGEFGDCFD', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGEGGGGGGGGGGGGGGGGGFGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFEC>CEGGGGGGGDEGFGDEGEGGGGGGGGGGGGGCF9D@', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGDEFFFGGFGGGFGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGDEFFGGGGGGGGGGFFGGGGCEEF?@FGGGGFGFGGG7DFGGGGGGFFFFGEC9EED', b'CCCCCGGDFFGGGFFEGGGGGGGGGGGGEGGGC@FFGG9CCEFGGGGGGGGG<FGEFDG:FGGGFGGGGG:@FFFGGDGGGFDGGG>7FFFGCGFGGGGGEGGGGFEFGGCD::FF=BFG88D=B+++,2>DG8,5;>+++4+,2,7', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFFGGGGFFGFGGGGGGGGGFGGGGGGFGGGGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGEFEEFDE@FGGCEE@E@CFEGGGGGFFGGECFGFGFCA', b'CCCCCFFGFGGGGGGGGGGGGGGGGCFEEGECGGGGGGGGGDCFGGG9AEFEFGGFFGGGG@FFFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGFEE>=BCFGFFGG?CEF9@8+8EGG8DFEFGGGGGGGFD,', ... b'CCCCCCFCCFGGCAECC@CGGGGGGGGGGF::FFCEFFEFGGFCFFDG<AEFECEGGGGGGGGE@<,CCFGG,EFGGGGGGGFGGGGC+EFG@FCFEFGGE,>ACF:4@F++8CCG+>7,++,:+++++,@,,@3@E**8>EF>E;D', b'CCCCCGGGGGGGFGGGGGGGGGGGGGGGGGGFD8FDGGGGFGFFGGGGGGGGGFFGGGGGGGGGGGGGGGGGGGGGGGGGGFFFGGGGGGGGGDFGEGGGGFEEDCEE>CFFC7>:CCC7=+4AE+CFG+,DD8F,@+@>>==D,EC', b'BBC9CAEGGGGDG;FCFF<FFGGFGGEGGEDEGCFF<;CFEFC:CD,CFFFFGFGCFGGGGGGGGGGGCEG@FFGGGGGGGGFGCGG+>FGGGGDFFCGFE8EFFAF@CEEEG@@>CCFC<FF@C+8+38@DDG<FEFFG83EECCC', b'CCCCCC@@CFEG8FFEDFFGGGGGEGCGGG>8CFEGCFFFGGFCFFGC@EFGGFF=E7FFGGGGCDFCFFGCGD9,CFEGEDEFGD=F+59?B?CF8BEFEFFFD<=+@=BCGGGD+@+@,8:=5@::@+@8>>F@F:D8,+23,,5', b'CCCCCDGGGGGGGGGGGGGGGGGGGDGDG7FGCGGGGFGGGGGGGFGGGGGGGFGGEGGGGGGFGGFFGGGGGFGFFGGGGGGGGGGGGGGGGGGGDGGGGCCG<ECCFF>C=BB=CEGG:EGGGDC6FGG@3BFF7@DFEFGD=F>', b'CCCCCGGGGGGGFF7@F,FEG<FCEGGGEGC7CFEG@F<FAFFFEGGGF:6EEA;,FF@FFGGGEE@GEFGGGG9?,,A,CFFGG+:CCGGGGCEFF@G<FDFD+,@8:>=7:+++B+>+:+@FE+8C3C,DE,>,>:DB,+7@:B;', b'CCCC@<<FGD<FGEGGFFCFFGGEGGGGGGGEFGGGGGGGGGGGGGGGGGGGGFFFC6FFGGGGGGFGGGG<FGFFGEEFGGGGECFBFFCFGDGGG9F<,,64=,,4+++4++@=+@@@,,,3,3++8+,87>F,8,33@,<E,,7', b'CCCCCGFDFFD@BFFFFFG9FGGGGGGGFGEFEFFGGFGDFFCFGFC@EEFFGG,@@FFEGGGGGEFGFFGGG9EDFGGGGGGGGGGGGGGGGGFECCGCFGGFEDFEE:CCEFGGD+6ACGG97?BF783,3,@CBC+6DA7>C=E', b'CCCCCGGGGGCFF@BFGGGFGGGGGGGG>FEDFFGFFGCGGGFGGGGGGGGGAEGFGC@FGGGGEFGGGDGGGGGFGGGGGGFGGGGGGGAFGGGGDGFGGFC?FFFCCGGEGGGC+CCFCGGGGGGC@838CE:FC>:>+,749@F', b'CCCCCGGGGGGGGEF@@FGGGGGGGGGGGGGGGGGGFFFGGGFGGGGGGFGFGGGGGGGGGGGGGGGAFGGGGGGGGGGGGGGGGGGGGGGDGGFFGGGFGGGGGF:=@FGEEFEECFGE:>@,5@>@@CFGGGF6D7>D8@CF7>;', b'CCCCCFGGGGGGGGGGFGGGGGGGFFFFGGGGGGGCFDFECFGGGGGFGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGFGGFCFFGFGEFGGFGGGGGEEEGGFGGEGC=DF@C@FFCFGFGFGGGGG7=@@GGE', b'CCCCC-@FE<CFGGGGC,FGGGEGGGGGGG:7CDFGGGGGGGGGGGGGGGGFFGF@ECFGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGGGGCFCF?+?BCFC:>CEFGGFCGGGGGGF+++>=<>,,:@=F@F+=@DF;@', b'CCCCCEFGGFGGFFG8FFFGGGGGGGGGGGFEFGEFG8CFGGGGGGFFGD8FCEGGGGGEGGGGGGGG@FGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDC,99>FEBC>B@+=CE,=FGG=CGECGGGCGEGCGGC6=@E9>', b'CCCCCFDEAFGGG7FDFFFFFGGGGGGGEGF@FDGGGGFFGGGGGGGGGFGFGGGGGGGGE7FFFFCFGGGGGG@FGGGDGD7,@FG7B,,:FGFCGFFEFGEE?,@7=FB@@=F7:C@F,+@>C+>:+3@D,@FGGGF?FDGGCG,', b'CCCCCEFFGGGGGGGGGGEGGGGGGCF@FC:FGGGC<CF<AECFEGCFGFEFGA@FE@@CCFDCFFFGGF7DEEFFGGGGCGFFGGGGGGGGGGCGFGFFFDF@,59AFG>4@+@C+@FFD@<CB3C<<CCC+5FFCC>CF8FF?E2', b'CCC-AFGGG<FCDFC,EFGEFF@CF@FFGC:+@<FF@FFCCEF@7FDGGGG<CFFGGCF7BFGGGGGDF9FGCE9F@FFGGGDDECFGDGGGEG<FFEGG:8ECF<?C:CECC+>+8@FG=CDGD++8>8=@8=FC8:@FF6,6=38', b'8B@A6-F-CC,CEFFGGGEDCFGEEGFF:CEDFFGF,CE@6BFGDFG@FFFC;A+8B,DF9EFFDB8FEF,CEG<@FF@FGF7+@>EFCEF6CFGCG@FFFGG,48@@@=>F48@>CCEFF=DFGGGADCFDF@C=,,8DDGC9D=3', b'CC<CCEFC,C<<@FGCFGEGGGGGGGGGFG7CFGGGGGCEFGFGGGG9C<FFDGGGGFEGGGGAE<FGFGGGGFGFG<FGGFCFGDGE+AC,CEGGEGGCGGG:7?;FGG7D9FBF+8++4++84@C++38+>8+3@F,,,8@B+3,', b'CCB9CFG9FEDG@F9<E,C<8<CCEDECC76@BFEGC8CA,CEFCDF<C@8,CAFF8CCEGGGGFEAFFEFFF,,9CFB9F@7CCF+:,,66B,,BBFDAF,94:+4+@BFFCCE+++63+,3::,3+8+633C,3,+463;:@73,'], dtype='|S147') - sequence_aligned(molecule)|S147b'CCCACCGCTCGGCTCAACTGGGTTTTCCCA...
array([b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGGGCGAGCGGGGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG--TTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTAAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTT-----GTTGAGCGCTTGCTAGGGTTTT-GCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGGGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGG--TTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', ... b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGGGCGGTGGG-----------------------------', b'CCCACCTCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTACCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCCGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGACGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCCGTAGCAGCGCGGGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAATTGCGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGTTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCTGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCT-CGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGT-GCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTCTCCCTAGCAAGCCGCTGCTCCGGTTTTCCGTAGCCGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGAAGCGCGAGCGGGGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCTAGTTGAGCGCTTGCTAGGATTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGG-TTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGATAAGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCATAGCAAGCCGATGCTACGGTTTTCCGTAGCGGCGCGAGCGGTTGG-----------------------------'], dtype='|S147') - sequence_quality_aligned(molecule)|S147b'GGGFGCF+@F7EGGGDFFGFGGCBFGGGFG...
array([b'GGGFGCF+@F7EGGGDFFGFGGCBFGGGFGGFGFGGGGGGGDGGGGGG<FGGGGGGGCEGGG<BF@CGGGGGGEFCECFGGGGCD<A+7>>+@44F+3@8 ', b'GGGFGGGGGGGGGGGGGGGGGFCFGGGG=EGFGGGGGGGGGFGGGGFGFGGGGGGGGGGGGFGGGGGGGGGGGG FEGGGGGGFEAAE@>EG7C=6:=F ', b'GGGGGGGGGGFEGGGGFGGGGGFGGGGGGGGFGGGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGECFFGGGGGGFGGGGGDFFGG@FGG ', b'GGGGGGGGGGECDCFFFGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCFGFFCFCGBC,9EFEGGGFFFGGDC@ECDEC74==CFFDEB ', b'GGGGGGGGGGEGGGGGGGGGGGFGGFEFFGGGGGGGGGGGEGGDFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEEEGGCFGG ', b'GGGGGGGEGGGGGGGGGGGGGGFBECFGGGGAFGGEFGGEGGGGGGGGGFGGGGGFGGGGGGCEFGGGGGGGGGGGGGFGGGGG7BEFE>@CEFGGCFGG ', b'GGGGGGGGFGC@FGGGGFGGGGFG7<FGEGGFFGGEAFGEEGGCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGEEBFC>:CEGGFEGG ', b'GGGGGGGGGG@CFGGGGGGGGGFGGGGGGFGFFGGFFFGGGEFCFGGGGGGGGGGGGGGGGGEFGGGGGGGGGG GGGGGGGDEGGGFCFGGEGGGFEEF ', b'GGGGGGGGGGC@EGGGGGGGGGFCG D=?FFF,CC@CDGAEEF?=B:F GAF@FGGFG7DEGCCEFFGGF,@7,<:FFCGGAFFGG4=F:CG7>F< ', b'GGGGGDGGGGEDGGGGGGCCFFFEGG9F@EEEEFGGGDECFGG9<FFGCFGFF<FGGGGGGGFEEGFFGGGGEEFGGGFGGGGGGF,=>==:=F:F@=FG ', b'GGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCGEGGGGGGGGGGG ', b'GGGGGGGGFGCGFFGFGGGGGGFGGG9FFGG<FGGGGGGGGGGGGGGGEFG?F@BEFGGGGDCGGEEGDGGGGGGGGGGGGGC8DFFFEC>=EGGGGGGE ', b'GGGGGGGGEEGGGDGGGGGGGG@FGGGGGGF<FFGGGGG>GFFGCGGFFFGGGGGGGGAFGGGGGDGGGGGGGG GGGGGGFGGGGFFDGGGGGGG=CEG ', b'GGGGGGGGGGGEGGGGGGGGGGFFGCGGGGGGFGGGGGGGGFGGGGGGEFGFGGGGGGGGGGGGGGGGGGGGGG8CGGGGEFGGGGFFCECCEFGGCFGE ', b'GGGGGGGGGGGGGGGGGGGGGGGDGFGFEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG GGGGGGGGGGGGFEGGGGGGGGD=F ', b'GGGGGGGGGGGGGGGGGGGGGFGEGGGGGGGGGGGGGGGGGFGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFEC>CEGGGGGGG ', b'GGGGGGGGGGGGGGGGGGGGGGFGDEFFFGGFGGGFGGGGGGGGGGFG GGGGGGGGGGGGGGGGGGGGGGDEFFGGGGGGGGGGFFGGGGCEEF?@FG ', b'GGGGGGGGEGGGC@FFGG9CCEFGGGGGGGGG<FGEFDG:FGGGFGGGGG:@FFFGGDGGGFDGGG>7FFFGCGFGGGGGEGGGGFEFGGCD::FF=BFG ', b'GGGGGGGGGGGGGGGGGGGGGGFFGGGGFFGFGGGGGGGGGFGGGGGGFGGGGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGEFEEFDE@FGG ', b'GGGGGCFEEGECGGGGGGGGGDCFGGG9AEFEFGGFFGGGG@FFFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGFEE>=BCFGFFGG ', ... b'GGGGGGGGGF::FFCEFFEFGGFCFFDG<AEFECEGGGGGGGGE@<,CCFGG,EFGGGGGGGFGGGGC+EFG@F CFEFGGE,>ACF:4@F++8CCG+>7 ', b'GGGGGGGGGGGFD8FDGGGGFGFFGGGGGGGGGFFGGGGGGGGGGGGGGGGGGGGGGGGGGFFFGGGGGGGGGDFGEGGGGFEEDCEE>CFFC7>:CCC7 ', b'FGGFGGEGGEDEGCFF<;CFEFC:CD,CFFFFGFGCFGGGGGGGGGGGCEG@FFGGGGGGGGFGCGG+>FGGGGDFFCGFE8EFFAF@CEEEG@@>CCFC ', b'GGGGEGCGGG>8CFEGCFFFGGFCFFGC@EFGGFF=E7FFGGGGCDFCFFGCGD9,CFEGEDEFGD=F+59?B?CF8BEFEFFFD<=+@=BCGGGD+@+@ ', b'GGGGGGDGDG7FGCGGGGFGGGGGGGFGGGGGGGFGGEGGGGGGFGGFFGGGGGFGFFGGGGGGGGGGGGGGGGGGGDGGGGCCG<ECCFF>C=BB=CEG ', b'G<FCEGGGEGC7CFEG@F<FAFFFEGGGF:6EEA;,FF@FFGGGEE@GEFGGGG9?,,A,CFFGG+:CCGGGGCEFF@G<FDFD+,@8:>=7:+++B+>+ ', b'FGGEGGGGGGGEFGGGGGGGGGGGGGGGGGGGGFFFC6FFGGGGGGFGGGG<FGFFGEEFGGGGECFBFFCFGDGGG9F<,,64=,,4+++4++@=+@@@ ', b'FGGGGGGGFGEFEFFGGFGDFFCFGFC@EEFFGG,@@FFEGGGGGEFGFFGGG9EDFGGGGGGGGGGGGGGGGG FECCGCFGGFEDFEE:CCEFGGD+6 ', b'GGGGGGGG>FEDFFGFFGCGGGFGGGGGGGGGAEGFGC@FGGGGEFGGGDGGGGGFGGGGGGFGGGGGGGAFGG GGDGFGGFC?FFFCCGGEGGGC+CC ', b'GGGGGGGGGGGGGGGGFFFGGGFGGGGGGFGFGGGGGGGGGGGGGGGAFGGGGGGGGGGGGGGGGGGGGGGDGGFFGGGFGGGGGF:=@FGEEFEECFGE ', b'GGGGFFFFGGGGGGGCFDFECFGGGGGFGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGG FGGFCFFGFGEFGGFGGGGGEEEGGFGGE ', b'GGEGGGGGGG:7CDFGGGGGGGGGGGGGGGGFFGF@ECFGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGGGGCFCF ?+?BCFC:>CEFGGFCG ', b'GGGGGGGGGGFEFGEFG8CFGGGGGGFFGD8FCEGGGGGEGGGGGGGG@FGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDC,99>FEBC>B@+=CE ', b'FGGGGGGGEGF@FDGGGGFFGGGGGGGGGFGFGGGGGGGGE7FFFFCFGGGGGG@FGGGDGD7,@FG7B,,:FGFCGFFEFGEE?,@7=FB@@=F7:C@F ', b'GGGGGCF@FC:FGGGC<CF<AECFEGCFGFEFGA@FE@@CCFDCFFFGGF7DEEFFGGGGCGFFGGGGGGGGGGCGFGFFFDF@,59AFG>4@+@C+@FF ', b'FF@CF@FFGC:+@<FF@FFCCEF@7FDGGGG<CFFGGCF7BFGGGGGDF9FGCE9F@FFGGGDDECFGDGGGEG<FFEGG:8ECF<?C:CECC+>+8@FG ', b'EDCFGEEGFF:CEDFFGF,CE@ 6BFGDFG@FFFC;A+8B,DF9EFFDB8FEF,CEG<@FF@FGF7+@>EFCEF6CFGCG@FFFGG,48@@@=>F48@>C ', b'GGGGGGGGFG7CFGGGGGCEFGFGGGG9C<FFDGGGGFEGGGGAE<FGFGGGGFGFG<FGGFCFGDGE+AC,CEGG:7?;FGG7D9FBF+8++4++84@C ', b'8<CCEDECC76@BFEGC8CA,CEFCDF<C@8,CAFF8CCEGGGGFEAFFEFFF,,9CFB9F@7CCF+:,,66B,,BBFDAF,94:+4+@BFFCCE+++63 '], dtype='|S147') - sequence_subset(molecule)|S8b'GGCGCCGC' ... b'GGCGCCGC'
array([b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', ... b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC'], dtype='|S8') - sequence_quality_subset(molecule)|S8b'7EGG<B7>' ... b'6@8C7C@B'
array([b'7EGG<B7>', b'GGGGGGE@', b'FEGGGGGG', b'ECGGFCEC', b'EGGGGGGG', b'GGFGCEE>', b'C@AFGGFC', b'@CFFEFCF', b'C@,C7DGG', b'EDGDFE>=', b'FFGGGGEG', b'CGGGCGEC', b'GGGGGGDG', b'GEGGGGCE', b'GGGGGGEG', b'GGGGGGEC', b'GGGGGGGG', b'GGFDDGGG', b'GGGGEGGE', b'ECFGGGEE', b'GGFGFG>>', b'+8GGFFGE', b'FBFFGFFG', b'BFGEGG=C', b'@@E8GG++', b'GFFGFGFG', b'GGGGGGBF', b'7+FEF7@:', b'FFFGGGFG', b'CCGGGG++', b'GGGGGGEG', b'FGGGGGFE', b'GGGGGGGG', b'C:F@FGDF', b'GGGGGG:E', b'FGGGGGGF', b'GCGGGFFG', b'GGGGGGGG', b'GGGGGGEG', b'@FGEGGCE', b'ECGGGGCG', b'FEGDFFDG', b'FEGEGG:E', b'7CGEFG@>', b'FEGGGGCE', b'F:GDGGGG', b'CFFGGG6@', b'CCFGGGGG', b'GGGCGGEG', b'EGFGGGGG', b'GGG@GGFG', b'GGFGGGGG', b'FEFE<FE7', b'CGGGGGGG', b'@@GGGGFG', b'EGGGGGGG', b'ECG:FGED', b'GGGGGF+8', b'@@FGCF++', b'GCGGGG+@', b'C@G:GF7C', b'++F:@E=@', b'+CFB,,>7', b'GGGGGGGG', b'FCFGGGGG', b'@6E@@<+>', b'CCF>GGEC', b'EEFC,D+8', b'FGGGGGGC', b'E:F@GG:>', b'GGGGGFGG', b'@@FEFGFE', b'CBGGGGFG', b'GGGGCFBE', b'GDFGFGGG', b'GGGGGGFG', b'@FFCGGDG', b'FECCCFBC', b'CFFGGG+>', b'DFGGGGGG', b'FGGGGGCG', b'+6A@4CFG', b'FGGDGGCE', b'CBGCFG7C', b'GGGGGGFE', b'GCDEGGFC', b'GGFGGGEG', b'GGGGGGGG', b'GGGGEGCC', b'GGGGGG:C', b'GGGGGGFG', b'GGGGGGGG', b'+6EFFGEG', b'FGGGFGCG', b'FGGGGGGG', b'CFGEGGFE', b'@6FEF<F6', b'EGGGGG=F', b'C@GGGGCF', b'GGGGGGDE', ... b'FGGGGGEG', b'CFFEGGGG', b'EGGFFG+C', b'GGFGFF:F', b'@CEF,CCF', b'CGGFGG7F', b'8FFEGGGG', b'GGGGGGCE', b'GGGGGGDG', b'FGGEGGCC', b'FEEEGG+4', b'E><@:,CF', b'GGFFFF:F', b'GGGGGGGG', b'GEGGGGGG', b'7:GCGG7F', b'CCCCGGGG', b'GEGGGGEG', b'C+FC7DEC', b'GFGEGGGG', b'GDGGGGCG', b'@+FC,C::', b'CCGCGFG+', b'FCFFFG7C', b'C@GGGG4A', b'++FF,6FF', b'+@F@<F+4', b'@:G@FFC+', b'GGGGGGEG', b'@FGGDGGG', b'+@GGF7:E', b':@GGFGFG', b'GGGGGGEG', b'FEFGGF+>', b'FEGDGG7F', b':@67?F++', b'E7ECFFFG', b'+6CF7F::', b'++C@FD+8', b'B:GFFF>F', b'C:FCFFCF', b'++,>DF=+', b'F6C@FGCC', b'C+;4GCG7', b'GFGGDFEG', b'GGFGGGFF', b'FFGGGGCG', b'+7FB4C>C', b':@GGGGFE', b'CDFFDG+@', b'++E7DF++', b'EGGGGECG', b'GGGGGGCG', b'@FEGGG4@', b'GFGGGGDG', b'GGEBGGC>', b'GGGGGGGG', b'+8GFGGCF', b'GGGGGGFG', b'C@GFGG:C', b'7:F:GGFF', b'CFGGGGGG', b'@:FFGGGG', b'F@GGFGEE', b'7CGGFGGC', b'GGGGGG@F', b'GGGGGGC@', b'GGECGGEG', b'FFFEGGC7', b'GGGGFD7B', b'CEEFGGEE', b'C@F:GF>F', b'GGE@7,FF', b'CEF7@F+=', b'GGGGGG@F', b'CCFC,DEG', b'C7G+,C++', b'+@GEGG9E', b':FDFD@C:', b'::GGFG4@', b'GFGGFF>C', b'DEFGFGCE', b'>8E7EF@=', b'7FGEGGCF', b'C7FFFG:>', b'GEC6GG++', b'EF@FGGEE', b'EDGCFGCC', b'GGGGGG@F', b'GGGGFGGG', b':7ECGGFC', b'FEGGGG>F', b'F@GG7,=F', b':FE@FFFG', b':+GCDD:C', b':CA+FG8@', b'7CGFCFF+', b'6@8C7C@B'], dtype='|S8') - sequence_coordinates(molecule, dimension)int646849 2670 9716 ... 11853 24880
array([[ 6849, 2670], [ 9716, 2565], [10664, 3332], ..., [14801, 23633], [14797, 24893], [11853, 24880]], dtype=int64) - selection_Cy5_active_start(molecule)boolFalse False True ... True False
array([False, False, True, True, True, False, False, False, False, False, False, False, True, True, False, True, False, False, True, False, False, False, True, True, False, False, False, False, False, True, True, True, False, False, False, False, True, False, True, True, True, False, True, False, True, True, True, True, True, False, False, False, False, True, False, False, False, True, True, False, True, True, True, True, True, True, False, True, False, True, False, True, True, True, True, False, True, False, True, False, True, False, True, True, False, True, False, False, False, False, True, False, False, False, False, True, True, False, True, False, True, True, True, False, True, True, True, True, True, False, True, True, True, False, True, True, False, False, False, False, True, True, False, False, True, True, True, False, True, True, False, True, True, True, True, True, False, True, True, True, False, False, True, True, True, True, False, False, False, True, True, True, True, True, False, False, False, False, True, True, False, False, False, True, True, True, True, False, True, True, False, False, False, False, False, False, True, True, True, False, ... False, True, True, True, False, False, True, False, True, False, False, True, False, False, True, False, True, True, True, True, True, False, False, False, False, True, True, True, False, False, False, False, True, True, True, False, False, True, True, True, True, True, False, False, False, False, False, False, True, True, False, True, True, False, True, True, False, False, True, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, True, True, False, True, True, False, True, True, False, False, True, False, False, False, False, False, True, False, True, True, True, False, False, False, True, True, False, True, True, True, True, True, False, True, False, False, True, False, False, False, False, True, True, True, True, False, False, True, True, True, False, False, False, True, False, True, True, False, False, False, True, True, False, True, True, True, True, False, False, True, True, True, False, True, True, True, False, False, False, False, False, True, False, True, False, False, True, True, True, True, False, False, True, True, True, True, False]) - selection_Cy5_active_end(molecule)boolFalse False True ... True False
array([False, False, True, True, True, False, False, False, False, False, False, False, True, True, False, True, False, False, True, False, False, False, True, False, False, False, False, False, False, False, True, False, False, False, False, False, True, False, True, True, True, False, True, False, True, True, True, True, True, False, False, False, False, True, False, False, False, True, True, False, False, True, False, True, True, True, False, True, False, True, False, True, True, True, True, False, True, False, True, False, True, False, True, True, False, False, False, False, False, False, True, False, False, False, False, True, True, False, True, False, False, True, True, False, True, False, True, True, False, False, True, True, True, False, True, True, False, False, False, False, True, False, False, False, True, True, True, False, True, False, False, True, True, True, True, True, False, False, True, True, False, False, True, True, True, True, False, False, False, True, True, True, True, True, False, False, False, True, False, True, False, False, False, True, True, True, True, False, True, True, False, False, False, False, False, False, True, True, True, False, ... False, True, True, True, False, False, False, False, True, False, False, False, False, False, False, False, True, True, True, True, False, False, False, False, False, True, True, True, False, False, False, False, False, True, True, False, False, True, True, False, True, True, False, False, False, False, False, False, True, True, False, True, True, False, True, True, False, False, True, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, False, True, False, True, True, False, True, False, False, False, True, False, False, False, False, False, False, False, False, True, False, False, False, False, True, True, False, True, True, True, False, True, False, False, False, False, True, False, False, False, False, False, False, True, True, False, False, True, True, True, False, False, False, False, False, True, True, False, False, False, True, True, False, True, False, True, True, False, False, True, True, True, False, True, True, True, False, False, False, False, False, True, False, True, False, False, True, True, True, True, False, False, True, True, True, True, False]) - selection_intensity_total(molecule)boolTrue True True ... True False True
array([ True, True, True, False, True, True, True, True, False, True, True, True, True, True, False, False, False, True, True, True, True, True, True, True, True, False, False, True, True, True, True, False, True, False, True, False, True, True, True, False, False, True, True, False, True, False, True, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, False, True, True, False, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, False, True, False, False, False, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, False, True, True, True, True, False, False, True, True, True, True, True, True, False, True, True, False, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, True, True, True, True, True, True, False, True, ... True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, True, False, False, False, True, True, True, True, True, True, True, False, True, True, False, False, True, True, True, True, True, False, True, True, False, True, False, False, False, False, False, False, True, True, False, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, False, True, True, True, True, False, True, True, True, True, True, False, False, True, True, True, False, True, False, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, False, False, False, True, False, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, False, True, True, True, False, True, False, True, True, True, False, True, True, False, False, True, False, True, True, True, True, False, True, True, False, True, False, True])
- channelPandasIndex
PandasIndex(Int64Index([0, 1], dtype='int64', name='channel'))
- dimensionPandasIndex
PandasIndex(Index([b'x', b'y'], dtype='object', name='dimension'))
- framePandasIndex
PandasIndex(Int64Index([ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, ... 390, 391, 392, 393, 394, 395, 396, 397, 398, 399], dtype='int64', name='frame', length=400))
Applying the selections changes the selected dataarray in the dataset.
[22]:
file.apply_selections(['selection_intensity_total', 'selection_Cy5_active_start', 'selection_Cy5_active_end'])
[23]:
file.selected
[23]:
<xarray.DataArray 'selected' (molecule: 753)>
array([False, False, True, False, True, False, False, False, False,
False, False, False, True, True, False, False, False, False,
True, False, False, False, True, False, False, False, False,
False, False, False, True, False, False, False, False, False,
True, False, True, False, False, False, True, False, True,
False, True, True, True, False, False, False, False, True,
False, False, False, True, True, False, False, True, False,
True, True, False, False, True, False, True, False, True,
True, True, True, False, True, False, False, False, True,
False, True, False, False, False, False, False, False, False,
True, False, False, False, False, True, True, False, False,
False, False, True, True, False, True, False, False, True,
False, False, True, True, True, False, True, False, False,
False, False, False, True, False, False, False, True, True,
True, False, True, False, False, True, True, True, False,
True, False, False, True, True, False, False, True, True,
True, False, False, False, False, True, True, True, True,
False, False, False, False, False, False, True, False, False,
False, True, True, True, True, False, True, False, False,
False, False, False, False, False, True, True, False, False,
...
False, True, True, True, False, False, False, False, True,
False, False, False, False, False, False, False, True, True,
True, False, False, False, False, False, False, True, True,
True, False, False, False, False, False, False, True, False,
False, True, True, False, True, True, False, False, False,
False, False, False, False, False, False, True, False, False,
True, True, False, False, False, True, False, False, False,
False, False, True, False, True, False, False, False, True,
False, True, True, True, False, True, True, True, False,
False, False, True, True, False, True, False, False, False,
False, False, False, False, False, False, False, False, False,
True, False, False, False, False, False, False, False, True,
True, False, False, False, False, False, False, False, True,
False, False, False, False, False, False, False, True, False,
False, True, True, False, False, False, False, False, False,
True, True, False, False, False, True, True, False, True,
False, True, True, False, False, False, True, True, False,
False, True, False, False, False, False, False, False, True,
False, False, False, False, True, True, True, True, False,
False, True, False, True, False, False])
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule
Attributes:
selection_names: ['selection_intensity_total', 'selection_Cy5_active_sta...- molecule: 753
- False False True False True False ... True False True False False
array([False, False, True, False, True, False, False, False, False, False, False, False, True, True, False, False, False, False, True, False, False, False, True, False, False, False, False, False, False, False, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, False, True, True, True, False, False, False, False, True, False, False, False, True, True, False, False, True, False, True, True, False, False, True, False, True, False, True, True, True, True, False, True, False, False, False, True, False, True, False, False, False, False, False, False, False, True, False, False, False, False, True, True, False, False, False, False, True, True, False, True, False, False, True, False, False, True, True, True, False, True, False, False, False, False, False, True, False, False, False, True, True, True, False, True, False, False, True, True, True, False, True, False, False, True, True, False, False, True, True, True, False, False, False, False, True, True, True, True, False, False, False, False, False, False, True, False, False, False, True, True, True, True, False, True, False, False, False, False, False, False, False, True, True, False, False, ... False, True, True, True, False, False, False, False, True, False, False, False, False, False, False, False, True, True, True, False, False, False, False, False, False, True, True, True, False, False, False, False, False, False, True, False, False, True, True, False, True, True, False, False, False, False, False, False, False, False, False, True, False, False, True, True, False, False, False, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, False, False, False, True, True, False, True, False, False, False, False, False, False, False, False, False, False, False, False, True, False, False, False, False, False, False, False, True, True, False, False, False, False, False, False, False, True, False, False, False, False, False, False, False, True, False, False, True, True, False, False, False, False, False, False, True, True, False, False, False, True, True, False, True, False, True, True, False, False, False, True, True, False, False, True, False, False, False, False, False, False, True, False, False, False, False, True, True, True, True, False, False, True, False, True, False, False]) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
- selection_names :
- ['selection_intensity_total', 'selection_Cy5_active_start', 'selection_Cy5_active_end']
Trace classification#
Here we classify the traces of selected molecules, i.e. we determine the state at each time point of the trace. First, we would like to exclude parts of the traces where the donor is inactive and where the total intensity is higher than the single-molecule threshold. Then we fit the remaining parts to one-state and two-state hidden Markov models (HMM) and determine which of the two fits best using the Bayesian nformation criterion (BIC).
Define classifications#
Determine where the donor is active.
[24]:
from scipy.signal import medfilt
[25]:
classification_donor_active = xr.DataArray(medfilt((file.intensity_total > intensity_total_min).astype(int), kernel_size=(1,11)).astype(bool),
name='classification_donor_active',
dims=('molecule','frame'))
classification_donor_active
[25]:
<xarray.DataArray 'classification_donor_active' (molecule: 753, frame: 400)>
array([[ True, True, True, ..., False, False, False],
[ True, True, True, ..., True, True, True],
[ True, True, True, ..., True, True, True],
...,
[ True, True, True, ..., True, True, True],
[ True, True, True, ..., True, True, True],
[ True, True, True, ..., True, True, True]])
Dimensions without coordinates: molecule, frame- molecule: 753
- frame: 400
- True True True True True True True ... True True True True True True
array([[ True, True, True, ..., False, False, False], [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], ..., [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True]])
Determin in which frames only a single dye is present.
[26]:
classification_single_dye = xr.DataArray(medfilt((file.intensity_total < intensity_total_max).astype(int), kernel_size=(1,11)).astype(bool),
name='classification_single_dye',
dims=('molecule','frame'))
classification_single_dye
[26]:
<xarray.DataArray 'classification_single_dye' (molecule: 753, frame: 400)>
array([[ True, True, True, ..., True, True, True],
[ True, True, True, ..., True, True, True],
[ True, True, True, ..., True, True, True],
...,
[ True, True, True, ..., True, True, True],
[False, False, False, ..., False, False, False],
[ True, True, True, ..., True, True, True]])
Dimensions without coordinates: molecule, frame- molecule: 753
- frame: 400
- True True True True True True True ... True True True True True True
array([[ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], ..., [ True, True, True, ..., True, True, True], [False, False, False, ..., False, False, False], [ True, True, True, ..., True, True, True]])
Save and apply classifications#
[27]:
file.set_variable(classification_donor_active)
file.set_variable(classification_single_dye)
The selection are now stored in the dataset for this sequence, starting with classification_:
[28]:
file.dataset
[28]:
<xarray.Dataset>
Dimensions: (molecule: 753, channel: 2, dimension: 2,
frame: 400)
Coordinates:
molecule_in_file (molecule) int32 999 719 141 ... 848 1340 352
file (molecule) |S58 b'Single-molecule data - gre...
* channel (channel) int64 0 1
* dimension (dimension) |S1 b'x' b'y'
time (frame) float64 0.0 0.123 0.245 ... 48.75 48.87
* frame (frame) int32 0 1 2 3 4 ... 395 396 397 398 399
illumination (frame) int32 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0
sequence_in_file (molecule) int32 386 412 949 ... 160 758 670
Dimensions without coordinates: molecule
Data variables: (12/21)
selected (molecule) bool False False ... False False
coordinates (molecule, channel, dimension) float64 24.92...
intensity (molecule, channel, frame) float64 1.559e+04...
intensity_raw (molecule, channel, frame) float64 9.597e+03...
FRET (molecule, frame) float64 0.0 0.0 ... 0.05922
intensity_red_before (molecule, channel) float64 5.718 ... -143.0
... ...
sequence_coordinates (molecule, dimension) int64 6849 2670 ... 24880
selection_Cy5_active_start (molecule) bool False False True ... True False
selection_Cy5_active_end (molecule) bool False False True ... True False
selection_intensity_total (molecule) bool True True True ... False True
classification_donor_active (molecule, frame) bool True True ... True True
classification_single_dye (molecule, frame) bool True True ... True True- molecule: 753
- channel: 2
- dimension: 2
- frame: 400
- molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - channel(channel)int640 1
array([0, 1], dtype=int64)
- dimension(dimension)|S1b'x' b'y'
array([b'x', b'y'], dtype='|S1')
- time(frame)float640.0 0.123 0.245 ... 48.75 48.87
- units :
- s
array([ 0. , 0.123, 0.245, 0.368, 0.49 , 0.612, 0.735, 0.858, 0.98 , 1.102, 1.225, 1.347, 1.47 , 1.592, 1.715, 1.838, 1.96 , 2.082, 2.205, 2.327, 2.45 , 2.573, 2.694, 2.817, 2.94 , 3.062, 3.185, 3.307, 3.429, 3.552, 3.675, 3.797, 3.92 , 4.042, 4.165, 4.287, 4.41 , 4.532, 4.654, 4.777, 4.9 , 5.022, 5.145, 5.267, 5.389, 5.512, 5.635, 5.757, 5.879, 6.002, 6.124, 6.247, 6.37 , 6.491, 6.614, 6.737, 6.859, 6.982, 7.104, 7.226, 7.349, 7.472, 7.594, 7.717, 7.839, 7.961, 8.084, 8.207, 8.329, 8.451, 8.574, 8.696, 8.819, 8.942, 9.063, 9.186, 9.309, 9.431, 9.553, 9.676, 9.799, 9.921, 10.044, 10.166, 10.288, 10.411, 10.534, 10.656, 10.778, 10.901, 11.024, 11.146, 11.269, 11.391, 11.513, 11.636, 11.759, 11.881, 12.003, 12.126, 12.248, 12.371, 12.494, 12.615, 12.738, 12.861, 12.983, 13.106, 13.228, 13.35 , 13.473, 13.596, 13.718, 13.841, 13.963, 14.085, 14.208, 14.331, 14.453, 14.575, 14.698, 14.82 , 14.943, 15.066, 15.187, 15.31 , 15.433, 15.555, 15.678, 15.8 , 15.922, 16.045, 16.168, 16.291, 16.413, 16.535, 16.658, 16.78 , 16.903, 17.026, 17.147, 17.27 , 17.393, 17.515, 17.638, 17.76 , 17.882, 18.005, 18.128, 18.25 , 18.372, 18.495, 18.618, 18.74 , 18.862, 18.985, 19.107, 19.23 , 19.353, 19.474, ... 29.396, 29.518, 29.64 , 29.763, 29.886, 30.008, 30.131, 30.253, 30.375, 30.498, 30.621, 30.743, 30.865, 30.988, 31.11 , 31.233, 31.356, 31.477, 31.6 , 31.723, 31.846, 31.968, 32.09 , 32.213, 32.335, 32.458, 32.581, 32.703, 32.825, 32.948, 33.07 , 33.193, 33.316, 33.437, 33.56 , 33.683, 33.805, 33.928, 34.05 , 34.172, 34.295, 34.418, 34.54 , 34.662, 34.785, 34.907, 35.03 , 35.153, 35.274, 35.397, 35.52 , 35.643, 35.765, 35.887, 36.01 , 36.132, 36.255, 36.378, 36.499, 36.622, 36.745, 36.867, 36.99 , 37.112, 37.234, 37.357, 37.48 , 37.602, 37.725, 37.847, 37.969, 38.092, 38.215, 38.337, 38.459, 38.582, 38.704, 38.827, 38.95 , 39.071, 39.194, 39.317, 39.439, 39.562, 39.685, 39.806, 39.929, 40.052, 40.174, 40.297, 40.419, 40.541, 40.664, 40.787, 40.909, 41.031, 41.154, 41.276, 41.399, 41.522, 41.644, 41.766, 41.889, 42.012, 42.134, 42.256, 42.379, 42.501, 42.624, 42.747, 42.868, 42.991, 43.114, 43.236, 43.359, 43.482, 43.603, 43.726, 43.849, 43.971, 44.094, 44.216, 44.338, 44.461, 44.584, 44.706, 44.828, 44.951, 45.073, 45.196, 45.319, 45.44 , 45.563, 45.686, 45.809, 45.931, 46.053, 46.176, 46.298, 46.421, 46.544, 46.665, 46.788, 46.911, 47.033, 47.156, 47.278, 47.4 , 47.523, 47.646, 47.768, 47.89 , 48.013, 48.135, 48.258, 48.381, 48.502, 48.625, 48.748, 48.87 ]) - frame(frame)int320 1 2 3 4 5 ... 395 396 397 398 399
array([ 0, 1, 2, ..., 397, 398, 399])
- illumination(frame)int320 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0
array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]) - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
- selected(molecule)boolFalse False True ... False False
- selection_names :
- ['selection_intensity_total', 'selection_Cy5_active_start', 'selection_Cy5_active_end']
array([False, False, True, False, True, False, False, False, False, False, False, False, True, True, False, False, False, False, True, False, False, False, True, False, False, False, False, False, False, False, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, False, True, True, True, False, False, False, False, True, False, False, False, True, True, False, False, True, False, True, True, False, False, True, False, True, False, True, True, True, True, False, True, False, False, False, True, False, True, False, False, False, False, False, False, False, True, False, False, False, False, True, True, False, False, False, False, True, True, False, True, False, False, True, False, False, True, True, True, False, True, False, False, False, False, False, True, False, False, False, True, True, True, False, True, False, False, True, True, True, False, True, False, False, True, True, False, False, True, True, True, False, False, False, False, True, True, True, True, False, False, False, False, False, False, True, False, False, False, True, True, True, True, False, True, False, False, False, False, False, False, False, True, True, False, False, ... False, True, True, True, False, False, False, False, True, False, False, False, False, False, False, False, True, True, True, False, False, False, False, False, False, True, True, True, False, False, False, False, False, False, True, False, False, True, True, False, True, True, False, False, False, False, False, False, False, False, False, True, False, False, True, True, False, False, False, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, False, False, False, True, True, False, True, False, False, False, False, False, False, False, False, False, False, False, False, True, False, False, False, False, False, False, False, True, True, False, False, False, False, False, False, False, True, False, False, False, False, False, False, False, True, False, False, True, True, False, False, False, False, False, False, True, True, False, False, False, True, True, False, True, False, True, True, False, False, False, True, True, False, False, True, False, False, False, False, False, False, True, False, False, False, False, True, True, True, True, False, False, True, False, True, False, False]) - coordinates(molecule, channel, dimension)float6424.92 341.5 283.8 ... 441.4 130.2
array([[[ 24.91602904, 341.48554967], [283.78574085, 339.137688 ]], [[ 78.17807134, 372.90395929], [336.78871819, 370.50780467]], [[ 92.01072464, 166.17974665], [350.74208901, 164.28798759]], ..., [[ 25.45838149, 469.78131449], [284.34290719, 467.1585397 ]], [[ 22.58199174, 129.18829072], [281.6534317 , 127.36065026]], [[183.17939012, 132.032136 ], [441.40299696, 130.15824268]]]) - intensity(molecule, channel, frame)float641.559e+04 1.252e+04 ... 884.1
- background_correction :
- [-300 -100]
- gamma_correction :
- 1.5
- alpha_correction :
- 0.05
array([[[ 1.55884625e+04, 1.25170638e+04, 1.25944564e+04, ..., 1.64734899e+02, 2.69480270e+02, -4.69745833e+02], [-1.14025092e+03, -1.79434893e+03, 7.10999508e+02, ..., -2.62304725e+03, -1.10312074e+03, -6.55675284e+02]], [[ 1.34390768e+04, 1.35416917e+04, 1.74872344e+04, ..., 1.56752700e+04, 1.63730120e+04, 1.66206681e+04], [-3.65498855e+01, -1.07613788e+03, 7.68370384e+02, ..., -1.29670147e+02, -6.38025128e+02, -2.18210334e+02]], [[ 1.15077644e+04, 9.18438669e+03, 9.73603353e+03, ..., 1.01456006e+04, 1.09333859e+04, 1.25990483e+04], [ 1.11070711e+04, 1.09403092e+04, 8.19838649e+03, ..., 6.23282590e+03, 1.21338662e+04, 1.14991815e+04]], ..., [[ 1.00809311e+04, 9.02672236e+03, 1.28988727e+04, ..., 1.20465479e+04, 1.01208656e+04, 9.69752167e+03], [ 4.66614740e+03, 7.48326565e+03, 5.04780621e+03, ..., 6.04748313e+03, 6.15144622e+03, 5.87020252e+03]], [[ 2.36006570e+04, 1.70832674e+04, 4.73061255e+04, ..., 1.34665923e+04, 1.30160141e+04, 1.55987137e+04], [ 1.41042367e+04, 1.63379632e+04, 7.00118411e+03, ..., 9.50686972e+03, 1.23659826e+04, 1.21960524e+04]], [[ 1.79517282e+04, 1.74761086e+04, 1.71619744e+04, ..., 1.65578237e+04, 1.52834714e+04, 1.40444913e+04], [ 1.54934599e+02, 5.60340529e+02, 2.93738407e+02, ..., 3.17724251e+01, -7.98358369e+02, 8.84103653e+02]]]) - intensity_raw(molecule, channel, frame)float649.597e+03 7.647e+03 ... 1.486e+03
array([[[ 9597.43649264, 7647.34209132, 7696.48025203, ..., -195.40641347, -128.90141607, -598.25132255], [ -460.82779502, -1268.49574232, 1240.72232826, ..., -2714.81050151, -1189.64672654, -779.16257571]], [[ 8232.74718725, 8297.89946185, 10803.00594487, ..., 9652.55236575, 10095.56319399, 10252.80514106], [ 535.40395552, -499.0533011 , 1542.73210166, ..., 554.09335198, 80.62547306, 512.82307128]], [[ 7006.51706311, 5531.35663118, 5881.60858793, ..., 6141.65119131, 6641.8323304 , 7699.39571645], [11582.45931601, 11299.52852699, 8585.18816608, ..., 6640.10592707, 12580.53554253, 12029.13387182]], ..., [[ 6100.59119767, 5431.2522946 , 7889.76041476, ..., 7348.60182896, 6125.94639233, 5857.15661519], [ 5070.19395799, 7834.60176685, 5592.74984693, ..., 6549.81052174, 6557.48949828, 6255.07860606]], [[14684.54410822, 10546.51897643, 29735.63522788, ..., 8250.21732206, 7964.13593817, 9603.94521165], [15184.26951661, 17092.12655598, 9266.49038866, ..., 10080.19933784, 12916.78333291, 12875.98811846]], [[11097.92266571, 10795.9419459 , 10596.49170729, ..., 10212.90391941, 9403.79135695, 8617.1373611 ], [ 952.52100907, 1334.14595752, 1051.83712911, ..., 759.6636088 , -134.1848 , 1486.32822049]]]) - FRET(molecule, frame)float640.0 0.0 0.05344 ... 0.0 0.05922
array([[0. , 0. , 0.05343669, ..., 1.06701138, 1.32325718, 0.58260439], [0. , 0. , 0.04208956, ..., 0. , 0. , 0. ], [0.49114092, 0.54362606, 0.4571314 , ..., 0.38055096, 0.52602131, 0.47717951], ..., [0.31641165, 0.45325688, 0.28126687, ..., 0.33422531, 0.37803149, 0.37707519], [0.37406913, 0.48884984, 0.1289179 , ..., 0.41381964, 0.48719503, 0.43878953], [0.00855677, 0.03106712, 0.01682764, ..., 0.0019152 , 0. , 0.05922216]]) - intensity_red_before(molecule, channel)float645.718 435.4 35.45 ... 76.4 -143.0
array([[ 5.71771351e+00, 4.35361883e+02], [ 3.54482215e+01, -1.41182975e+02], [-2.72161708e+01, 7.28438296e+03], ..., [ 6.17814544e+01, 4.85823584e+03], [ 1.73779796e+01, 6.37530474e+03], [ 7.64042010e+01, -1.43028955e+02]]) - intensity_red_after(molecule, channel)float6449.42 103.4 -14.57 ... 13.65 111.7
array([[ 49.42475415, 103.44932566], [ -14.57350384, 244.16745202], [ 21.27267505, 6472.26269457], ..., [ 52.97241636, 4773.01440448], [ 60.07183329, 5599.77061963], [ 13.64937453, 111.68762698]]) - sequence_name(molecule)|S10b'HJ_general' ... b'HJ_general'
array([b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', ... b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general', b'HJ_general'], dtype='|S10') - sequence_tile(molecule)int641101 1101 1101 ... 1102 1102 1102
array([1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, 1101, ... 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102, 1102], dtype=int64) - sequence(molecule)|S147b'TACCATCCCAGGCATGTCCACCCACCGCTC...
array([b'TACCATCCCAGGCATGTCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGGGCGAGCGGGGGGAGATAGGAAGAGCAGATTCTGAACTAC', b'TACCATGCTCGCCAGCTCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCCA', b'TACCAATCAAGTGATGTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTAAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCACACCTGGGACATCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACT', b'TACCAGGCGAGGAATGTATACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCATACCTGCAAGTTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCACCCCAGTTACCTAGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAAATCGGAAGAGCACACGTCTGAACTC', b'TACCAGACCTGTGAAGTCCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCAGCCTTGGCACGTCCACCCACCGCTCGGCTCAACTGGGTTTGTTGAGCGCTTGCTAGGGTTTTGCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGGGGGAGATCGGAAGAGCACACGTCTGAACTCCAGTCA', b'TACCAGGCCGGTCATATTAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAAACTGGCTAATTGTACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAACCCGGATACCTAGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAGACAGGGCACCTTGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCAATCCGGTGAGTTAAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCACCCGTGTGATTTTGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCACACGCGCTAAGTCGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAGCCAAGACATCTCAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCCA', b'TGTTTAGTAACGTACGCGCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAAACGAACTCGTATGCCGTCCTCTGCT', b'TACCACTCCAGCAATTTGCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAAGCAGGTAAAATATACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', ... b'TACCAACCGAGGTAATTCTACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGGGCGGTGGGAGATCGGAAGAGCAGCCGTCTGAACTCC', b'TACCATCCACGCCATCTCCACCCACCTCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAAATC', b'TACCAACCACGGGAGTTTTACCCACCGCTCGGCTCAACTGGGTTTTACCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCATGCTCGTGAGCTTAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCCGCGCGAGCGGTGGGAGATCGGAAGAGAACACGTCTTAACTC', b'TACAATCACGTGAGTTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCA', b'TACCATCCACGCGAATTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGACGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAAATC', b'TACCAAACTTGTCAACTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCCGTAGCAGCGCGGGCGGTGGGAAATCGGAAGAGCACACTTCTGAACTC', b'TACCAGACCGGATATCTTCACCCACCGCTCGGCTCAACTGGGTTTTCCCAATTGCGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCACTCTTGCGAACTTCACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGCGCACACGTCTGAACTCC', b'TACCAGTCCCGCGATCTTGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGTTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCAGACAGGACATGTTTACCCACCGCTCGGCTCAACTGGGTTTTCCCTGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTCGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCATTCGGGACACTTGAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTCC', b'TACCAAGCACGGTAGGTTAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCATTCTGGGGATATCAACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTCTCCCTAGCAAGCCGCTGCTCCGGTTTTCCGTAGCCGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTT', b'TACCATACGTGTGACTTACACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGAAGCGCGAGCGGGGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACAAGCCCAGTAATTTAGACCCACCGCTCGGCTCAACTGGGTTTTCCTAGTTGAGCGCTTGCTAGGATTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTCTGAACTC', b'TACCCCATGTCAGCTAAACCCACCGCTCGGCTCAACTGGGTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGGAGATCGGAAGAGCACACGTATGAACTACAG', b'TACCAACCCTGCCATGTGTACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGATAAGGTTCTACGGTTTCCGTAGCAGCGCGAGCGGTGGGCGATCGGAAGAGCACACGTT', b'TACCATGCCAGCTAGGTGGACCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCATAGCAAGCCGATGCTACGGTTTTCCGTAGCGGCGCGAGCGGTTGGGAATCGGGTGAGCAAACGTCTGAACTT'], dtype='|S147') - sequence_quality(molecule)|S147b'AB@<6-C,6CFGCGGGGCGGGGGFGCF+@F...
array([b'AB@<6-C,6CFGCGGGGCGGGGGFGCF+@F7EGGGDFFGFGGCBFGGGFGGFGFGGGGGGGDGGGGGG<FGGGGGGGCEGGG<BF@CGGGGGGEFCECFGGGGCD<A+7>>+@44F+3@8@78:,=C:+*42=<*++,75B<*00<<', b'CCCCCGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGGGGGGFCFGGGG=EGFGGGGGGGGGFGGGGFGFGGGGGGGGGGGGFGGGGGGGGGGGGFEGGGGGGFEAAE@>EG7C=6:=F@ECDFFFDE:=DFFFDGG@@FD=E=<<E3', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGFEGGGGFGGGGGFGGGGGGGGFGGGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGECFFGGGGGGFGGGGGDFFGG@FGGCEGG98>=B@ECEGGGGGFGGFGGF83', b'CCCCCGGGGGEFFGGGGGGGGGGGGGGGGGECDCFFFGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCFGFFCFCGBC,9EFEGGGFFFGGDC@ECDEC74==CFFDEBDE7,2@C7@FFGD8EGDC8+8?FFG7', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGGGGFGGFEFFGGGGGGGGGGGEGGDFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEEEGGCFGGFCCEGDEGEGGGGGGGGFBF8:FGEF9', b'CCCCCGGGGGGGGGGFFGGGGGGGGGGEGGGGGGGGGGGGGGFBECFGGGGAFGGEFGGEGGGGGGGGGFGGGGGFGGGGGGCEFGGGGGGGGGGGGGFGGGGG7BEFE>@CEFGGCFGGGGG8ED>CCEFGGGGGGC=DF8DFD,3', b'CCCCCGGGGGGGFAFFGGGGGGGGGGGGFGC@FGGGGFGGGGFG7<FGEGGFFGGEAFGEEGGCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGEEBFC>:CEGGFEGGFGFGF@CFBCFFGGGGFGFFFFGG8==', b'@CCCCDGDGGFFFFGGFGGFGGGGGGGGGG@CFGGGGGGGGGFGGGGGGFGFFGGFFFGGGEFCFGGGGGGGGGGGGGGGGGEFGGGGGGGGGGGGGGGGGDEGGGFCFGGEGGGFEEFGGE,CF@CFEGDD@=DFGGGGFFGD@EE', b'CCCCCFDFFGFGEGFDGG@FGGGGGGGGGGC@EGGGGGGGGGFCGD=?FFF,CC@CDGAEEF?=B:FGAF@FGGFG7DEGCCEFFGGF,@7,<:FFCGGAFFGG4=F:CG7>F<:FE+2BFDEGG76FGFCE@**;AFC00<>+<C0', b'CCCCCGGGEFGGGGGGGGGGGGGGGDGGGGEDGGGGGGCCFFFEGG9F@EEEEFGGGDECFGG9<FFGCFGFF<FGGGGGGGFEEGFFGGGGEEFGGGFGGGGGGF,=>==:=F:F@=FG?CEFE@=FE>DGFGCGGFDFC4CFGC,', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCGEGGGGGGGGGGGDCGGGDCE8CFGGGGGGCFGGFDGEEA', b'CCCCCGGGGGDGEGGGGGGGGGGGGGGGFGCGFFGFGGGGGGFGGG9FFGG<FGGGGGGGGGGGGGGGEFG?F@BEFGGGGDCGGEEGDGGGGGGGGGGGGGC8DFFFEC>=EGGGGGGE88EFFDCGEGGFFGGFFFFCF4+@@E;', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGEEGGGDGGGGGGGG@FGGGGGGF<FFGGGGG>GFFGCGGFFFGGGGGGGGAFGGGGGDGGGGGGGGGGGGGGFGGGGFFDGGGGGGG=CEGGGG,CFCGGDFF,@8DFG8D8DEF?DFE', b'CCCCCGFFGGGGGGGFGGGGGGGGGGGGGGGEGGGGGGGGGGFFGCGGGGGGFGGGGGGGGFGGGGGGEFGFGGGGGGGGGGGGGGGGGGGGGG8CGGGGEFGGGGFFCECCEFGGCFGE:>=,C6@FEEGGGGGF?F=EFFDGAA,', b'CCCCCGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGFGFEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFEGGGGGGGGD=FGGGGGGGGGGGGGFGGGGGGGEFGDCFD', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGEGGGGGGGGGGGGGGGGGFGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFEC>CEGGGGGGGDEGFGDEGEGGGGGGGGGGGGGCF9D@', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFGDEFFFGGFGGGFGGGGGGGGGGFGGGGGGGGGGGGGGGGGGGGGGGDEFFGGGGGGGGGGFFGGGGCEEF?@FGGGGFGFGGG7DFGGGGGGFFFFGEC9EED', b'CCCCCGGDFFGGGFFEGGGGGGGGGGGGEGGGC@FFGG9CCEFGGGGGGGGG<FGEFDG:FGGGFGGGGG:@FFFGGDGGGFDGGG>7FFFGCGFGGGGGEGGGGFEFGGCD::FF=BFG88D=B+++,2>DG8,5;>+++4+,2,7', b'CCCCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFFGGGGFFGFGGGGGGGGGFGGGGGGFGGGGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGEFEEFDE@FGGCEE@E@CFEGGGGGFFGGECFGFGFCA', b'CCCCCFFGFGGGGGGGGGGGGGGGGCFEEGECGGGGGGGGGDCFGGG9AEFEFGGFFGGGG@FFFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGFEE>=BCFGFFGG?CEF9@8+8EGG8DFEFGGGGGGGFD,', ... b'CCCCCCFCCFGGCAECC@CGGGGGGGGGGF::FFCEFFEFGGFCFFDG<AEFECEGGGGGGGGE@<,CCFGG,EFGGGGGGGFGGGGC+EFG@FCFEFGGE,>ACF:4@F++8CCG+>7,++,:+++++,@,,@3@E**8>EF>E;D', b'CCCCCGGGGGGGFGGGGGGGGGGGGGGGGGGFD8FDGGGGFGFFGGGGGGGGGFFGGGGGGGGGGGGGGGGGGGGGGGGGGFFFGGGGGGGGGDFGEGGGGFEEDCEE>CFFC7>:CCC7=+4AE+CFG+,DD8F,@+@>>==D,EC', b'BBC9CAEGGGGDG;FCFF<FFGGFGGEGGEDEGCFF<;CFEFC:CD,CFFFFGFGCFGGGGGGGGGGGCEG@FFGGGGGGGGFGCGG+>FGGGGDFFCGFE8EFFAF@CEEEG@@>CCFC<FF@C+8+38@DDG<FEFFG83EECCC', b'CCCCCC@@CFEG8FFEDFFGGGGGEGCGGG>8CFEGCFFFGGFCFFGC@EFGGFF=E7FFGGGGCDFCFFGCGD9,CFEGEDEFGD=F+59?B?CF8BEFEFFFD<=+@=BCGGGD+@+@,8:=5@::@+@8>>F@F:D8,+23,,5', b'CCCCCDGGGGGGGGGGGGGGGGGGGDGDG7FGCGGGGFGGGGGGGFGGGGGGGFGGEGGGGGGFGGFFGGGGGFGFFGGGGGGGGGGGGGGGGGGGDGGGGCCG<ECCFF>C=BB=CEGG:EGGGDC6FGG@3BFF7@DFEFGD=F>', b'CCCCCGGGGGGGFF7@F,FEG<FCEGGGEGC7CFEG@F<FAFFFEGGGF:6EEA;,FF@FFGGGEE@GEFGGGG9?,,A,CFFGG+:CCGGGGCEFF@G<FDFD+,@8:>=7:+++B+>+:+@FE+8C3C,DE,>,>:DB,+7@:B;', b'CCCC@<<FGD<FGEGGFFCFFGGEGGGGGGGEFGGGGGGGGGGGGGGGGGGGGFFFC6FFGGGGGGFGGGG<FGFFGEEFGGGGECFBFFCFGDGGG9F<,,64=,,4+++4++@=+@@@,,,3,3++8+,87>F,8,33@,<E,,7', b'CCCCCGFDFFD@BFFFFFG9FGGGGGGGFGEFEFFGGFGDFFCFGFC@EEFFGG,@@FFEGGGGGEFGFFGGG9EDFGGGGGGGGGGGGGGGGGFECCGCFGGFEDFEE:CCEFGGD+6ACGG97?BF783,3,@CBC+6DA7>C=E', b'CCCCCGGGGGCFF@BFGGGFGGGGGGGG>FEDFFGFFGCGGGFGGGGGGGGGAEGFGC@FGGGGEFGGGDGGGGGFGGGGGGFGGGGGGGAFGGGGDGFGGFC?FFFCCGGEGGGC+CCFCGGGGGGC@838CE:FC>:>+,749@F', b'CCCCCGGGGGGGGEF@@FGGGGGGGGGGGGGGGGGGFFFGGGFGGGGGGFGFGGGGGGGGGGGGGGGAFGGGGGGGGGGGGGGGGGGGGGGDGGFFGGGFGGGGGF:=@FGEEFEECFGE:>@,5@>@@CFGGGF6D7>D8@CF7>;', b'CCCCCFGGGGGGGGGGFGGGGGGGFFFFGGGGGGGCFDFECFGGGGGFGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGFGGFCFFGFGEFGGFGGGGGEEEGGFGGEGC=DF@C@FFCFGFGFGGGGG7=@@GGE', b'CCCCC-@FE<CFGGGGC,FGGGEGGGGGGG:7CDFGGGGGGGGGGGGGGGGFFGF@ECFGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGGGGCFCF?+?BCFC:>CEFGGFCGGGGGGF+++>=<>,,:@=F@F+=@DF;@', b'CCCCCEFGGFGGFFG8FFFGGGGGGGGGGGFEFGEFG8CFGGGGGGFFGD8FCEGGGGGEGGGGGGGG@FGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDC,99>FEBC>B@+=CE,=FGG=CGECGGGCGEGCGGC6=@E9>', b'CCCCCFDEAFGGG7FDFFFFFGGGGGGGEGF@FDGGGGFFGGGGGGGGGFGFGGGGGGGGE7FFFFCFGGGGGG@FGGGDGD7,@FG7B,,:FGFCGFFEFGEE?,@7=FB@@=F7:C@F,+@>C+>:+3@D,@FGGGF?FDGGCG,', b'CCCCCEFFGGGGGGGGGGEGGGGGGCF@FC:FGGGC<CF<AECFEGCFGFEFGA@FE@@CCFDCFFFGGF7DEEFFGGGGCGFFGGGGGGGGGGCGFGFFFDF@,59AFG>4@+@C+@FFD@<CB3C<<CCC+5FFCC>CF8FF?E2', b'CCC-AFGGG<FCDFC,EFGEFF@CF@FFGC:+@<FF@FFCCEF@7FDGGGG<CFFGGCF7BFGGGGGDF9FGCE9F@FFGGGDDECFGDGGGEG<FFEGG:8ECF<?C:CECC+>+8@FG=CDGD++8>8=@8=FC8:@FF6,6=38', b'8B@A6-F-CC,CEFFGGGEDCFGEEGFF:CEDFFGF,CE@6BFGDFG@FFFC;A+8B,DF9EFFDB8FEF,CEG<@FF@FGF7+@>EFCEF6CFGCG@FFFGG,48@@@=>F48@>CCEFF=DFGGGADCFDF@C=,,8DDGC9D=3', b'CC<CCEFC,C<<@FGCFGEGGGGGGGGGFG7CFGGGGGCEFGFGGGG9C<FFDGGGGFEGGGGAE<FGFGGGGFGFG<FGGFCFGDGE+AC,CEGGEGGCGGG:7?;FGG7D9FBF+8++4++84@C++38+>8+3@F,,,8@B+3,', b'CCB9CFG9FEDG@F9<E,C<8<CCEDECC76@BFEGC8CA,CEFCDF<C@8,CAFF8CCEGGGGFEAFFEFFF,,9CFB9F@7CCF+:,,66B,,BBFDAF,94:+4+@BFFCCE+++63+,3::,3+8+633C,3,+463;:@73,'], dtype='|S147') - sequence_aligned(molecule)|S147b'CCCACCGCTCGGCTCAACTGGGTTTTCCCA...
array([b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGGGCGAGCGGGGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG--TTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTAAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTT-----GTTGAGCGCTTGCTAGGGTTTT-GCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGGGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGG--TTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', ... b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGGGCGGTGGG-----------------------------', b'CCCACCTCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTACCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCCGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGACGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTCCCGTAGCAGCGCGGGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAATTGCGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGG-TTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGTTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCTGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCT-CGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGT-GCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTCTCCCTAGCAAGCCGCTGCTCCGGTTTTCCGTAGCCGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGAAGCGCGAGCGGGGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCTAGTTGAGCGCTTGCTAGGATTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGG-TTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGCTACGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCCTAGCAAGCCGCTGATAAGGTTTTCCGTAGCAGCGCGAGCGGTGGG-----------------------------', b'CCCACCGCTCGGCTCAACTGGGTTTTCCCAGTTGAGCGCTTGCTAGGGTTTTCCATAGCAAGCCGATGCTACGGTTTTCCGTAGCGGCGCGAGCGGTTGG-----------------------------'], dtype='|S147') - sequence_quality_aligned(molecule)|S147b'GGGFGCF+@F7EGGGDFFGFGGCBFGGGFG...
array([b'GGGFGCF+@F7EGGGDFFGFGGCBFGGGFGGFGFGGGGGGGDGGGGGG<FGGGGGGGCEGGG<BF@CGGGGGGEFCECFGGGGCD<A+7>>+@44F+3@8 ', b'GGGFGGGGGGGGGGGGGGGGGFCFGGGG=EGFGGGGGGGGGFGGGGFGFGGGGGGGGGGGGFGGGGGGGGGGGG FEGGGGGGFEAAE@>EG7C=6:=F ', b'GGGGGGGGGGFEGGGGFGGGGGFGGGGGGGGFGGGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGECFFGGGGGGFGGGGGDFFGG@FGG ', b'GGGGGGGGGGECDCFFFGGGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCFGFFCFCGBC,9EFEGGGFFFGGDC@ECDEC74==CFFDEB ', b'GGGGGGGGGGEGGGGGGGGGGGFGGFEFFGGGGGGGGGGGEGGDFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGEEEGGCFGG ', b'GGGGGGGEGGGGGGGGGGGGGGFBECFGGGGAFGGEFGGEGGGGGGGGGFGGGGGFGGGGGGCEFGGGGGGGGGGGGGFGGGGG7BEFE>@CEFGGCFGG ', b'GGGGGGGGFGC@FGGGGFGGGGFG7<FGEGGFFGGEAFGEEGGCCGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDGGGGGGGEEBFC>:CEGGFEGG ', b'GGGGGGGGGG@CFGGGGGGGGGFGGGGGGFGFFGGFFFGGGEFCFGGGGGGGGGGGGGGGGGEFGGGGGGGGGG GGGGGGGDEGGGFCFGGEGGGFEEF ', b'GGGGGGGGGGC@EGGGGGGGGGFCG D=?FFF,CC@CDGAEEF?=B:F GAF@FGGFG7DEGCCEFFGGF,@7,<:FFCGGAFFGG4=F:CG7>F< ', b'GGGGGDGGGGEDGGGGGGCCFFFEGG9F@EEEEFGGGDECFGG9<FFGCFGFF<FGGGGGGGFEEGFFGGGGEEFGGGFGGGGGGF,=>==:=F:F@=FG ', b'GGGGGGGGGGFFFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGCGEGGGGGGGGGGG ', b'GGGGGGGGFGCGFFGFGGGGGGFGGG9FFGG<FGGGGGGGGGGGGGGGEFG?F@BEFGGGGDCGGEEGDGGGGGGGGGGGGGC8DFFFEC>=EGGGGGGE ', b'GGGGGGGGEEGGGDGGGGGGGG@FGGGGGGF<FFGGGGG>GFFGCGGFFFGGGGGGGGAFGGGGGDGGGGGGGG GGGGGGFGGGGFFDGGGGGGG=CEG ', b'GGGGGGGGGGGEGGGGGGGGGGFFGCGGGGGGFGGGGGGGGFGGGGGGEFGFGGGGGGGGGGGGGGGGGGGGGG8CGGGGEFGGGGFFCECCEFGGCFGE ', b'GGGGGGGGGGGGGGGGGGGGGGGDGFGFEGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGG GGGGGGGGGGGGFEGGGGGGGGD=F ', b'GGGGGGGGGGGGGGGGGGGGGFGEGGGGGGGGGGGGGGGGGFGGFGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGFEC>CEGGGGGGG ', b'GGGGGGGGGGGGGGGGGGGGGGFGDEFFFGGFGGGFGGGGGGGGGGFG GGGGGGGGGGGGGGGGGGGGGGDEFFGGGGGGGGGGFFGGGGCEEF?@FG ', b'GGGGGGGGEGGGC@FFGG9CCEFGGGGGGGGG<FGEFDG:FGGGFGGGGG:@FFFGGDGGGFDGGG>7FFFGCGFGGGGGEGGGGFEFGGCD::FF=BFG ', b'GGGGGGGGGGGGGGGGGGGGGGFFGGGGFFGFGGGGGGGGGFGGGGGGFGGGGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGGGGEFEEFDE@FGG ', b'GGGGGCFEEGECGGGGGGGGGDCFGGG9AEFEFGGFFGGGG@FFFGGGFGGGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGGGGGGGFEE>=BCFGFFGG ', ... b'GGGGGGGGGF::FFCEFFEFGGFCFFDG<AEFECEGGGGGGGGE@<,CCFGG,EFGGGGGGGFGGGGC+EFG@F CFEFGGE,>ACF:4@F++8CCG+>7 ', b'GGGGGGGGGGGFD8FDGGGGFGFFGGGGGGGGGFFGGGGGGGGGGGGGGGGGGGGGGGGGGFFFGGGGGGGGGDFGEGGGGFEEDCEE>CFFC7>:CCC7 ', b'FGGFGGEGGEDEGCFF<;CFEFC:CD,CFFFFGFGCFGGGGGGGGGGGCEG@FFGGGGGGGGFGCGG+>FGGGGDFFCGFE8EFFAF@CEEEG@@>CCFC ', b'GGGGEGCGGG>8CFEGCFFFGGFCFFGC@EFGGFF=E7FFGGGGCDFCFFGCGD9,CFEGEDEFGD=F+59?B?CF8BEFEFFFD<=+@=BCGGGD+@+@ ', b'GGGGGGDGDG7FGCGGGGFGGGGGGGFGGGGGGGFGGEGGGGGGFGGFFGGGGGFGFFGGGGGGGGGGGGGGGGGGGDGGGGCCG<ECCFF>C=BB=CEG ', b'G<FCEGGGEGC7CFEG@F<FAFFFEGGGF:6EEA;,FF@FFGGGEE@GEFGGGG9?,,A,CFFGG+:CCGGGGCEFF@G<FDFD+,@8:>=7:+++B+>+ ', b'FGGEGGGGGGGEFGGGGGGGGGGGGGGGGGGGGFFFC6FFGGGGGGFGGGG<FGFFGEEFGGGGECFBFFCFGDGGG9F<,,64=,,4+++4++@=+@@@ ', b'FGGGGGGGFGEFEFFGGFGDFFCFGFC@EEFFGG,@@FFEGGGGGEFGFFGGG9EDFGGGGGGGGGGGGGGGGG FECCGCFGGFEDFEE:CCEFGGD+6 ', b'GGGGGGGG>FEDFFGFFGCGGGFGGGGGGGGGAEGFGC@FGGGGEFGGGDGGGGGFGGGGGGFGGGGGGGAFGG GGDGFGGFC?FFFCCGGEGGGC+CC ', b'GGGGGGGGGGGGGGGGFFFGGGFGGGGGGFGFGGGGGGGGGGGGGGGAFGGGGGGGGGGGGGGGGGGGGGGDGGFFGGGFGGGGGF:=@FGEEFEECFGE ', b'GGGGFFFFGGGGGGGCFDFECFGGGGGFGGGGGGGGGGEGGGGGGGGGGGGGGGGGGGGGGGFGGGGGGG FGGFCFFGFGEFGGFGGGGGEEEGGFGGE ', b'GGEGGGGGGG:7CDFGGGGGGGGGGGGGGGGFFGF@ECFGGGGGGGGGGGGGGGGGGGGGGGGGGGEGGGGGGGGGGGCFCF ?+?BCFC:>CEFGGFCG ', b'GGGGGGGGGGFEFGEFG8CFGGGGGGFFGD8FCEGGGGGEGGGGGGGG@FGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGGDC,99>FEBC>B@+=CE ', b'FGGGGGGGEGF@FDGGGGFFGGGGGGGGGFGFGGGGGGGGE7FFFFCFGGGGGG@FGGGDGD7,@FG7B,,:FGFCGFFEFGEE?,@7=FB@@=F7:C@F ', b'GGGGGCF@FC:FGGGC<CF<AECFEGCFGFEFGA@FE@@CCFDCFFFGGF7DEEFFGGGGCGFFGGGGGGGGGGCGFGFFFDF@,59AFG>4@+@C+@FF ', b'FF@CF@FFGC:+@<FF@FFCCEF@7FDGGGG<CFFGGCF7BFGGGGGDF9FGCE9F@FFGGGDDECFGDGGGEG<FFEGG:8ECF<?C:CECC+>+8@FG ', b'EDCFGEEGFF:CEDFFGF,CE@ 6BFGDFG@FFFC;A+8B,DF9EFFDB8FEF,CEG<@FF@FGF7+@>EFCEF6CFGCG@FFFGG,48@@@=>F48@>C ', b'GGGGGGGGFG7CFGGGGGCEFGFGGGG9C<FFDGGGGFEGGGGAE<FGFGGGGFGFG<FGGFCFGDGE+AC,CEGG:7?;FGG7D9FBF+8++4++84@C ', b'8<CCEDECC76@BFEGC8CA,CEFCDF<C@8,CAFF8CCEGGGGFEAFFEFFF,,9CFB9F@7CCF+:,,66B,,BBFDAF,94:+4+@BFFCCE+++63 '], dtype='|S147') - sequence_subset(molecule)|S8b'GGCGCCGC' ... b'GGCGCCGC'
array([b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', ... b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC', b'GGCGCCGC'], dtype='|S8') - sequence_quality_subset(molecule)|S8b'7EGG<B7>' ... b'6@8C7C@B'
array([b'7EGG<B7>', b'GGGGGGE@', b'FEGGGGGG', b'ECGGFCEC', b'EGGGGGGG', b'GGFGCEE>', b'C@AFGGFC', b'@CFFEFCF', b'C@,C7DGG', b'EDGDFE>=', b'FFGGGGEG', b'CGGGCGEC', b'GGGGGGDG', b'GEGGGGCE', b'GGGGGGEG', b'GGGGGGEC', b'GGGGGGGG', b'GGFDDGGG', b'GGGGEGGE', b'ECFGGGEE', b'GGFGFG>>', b'+8GGFFGE', b'FBFFGFFG', b'BFGEGG=C', b'@@E8GG++', b'GFFGFGFG', b'GGGGGGBF', b'7+FEF7@:', b'FFFGGGFG', b'CCGGGG++', b'GGGGGGEG', b'FGGGGGFE', b'GGGGGGGG', b'C:F@FGDF', b'GGGGGG:E', b'FGGGGGGF', b'GCGGGFFG', b'GGGGGGGG', b'GGGGGGEG', b'@FGEGGCE', b'ECGGGGCG', b'FEGDFFDG', b'FEGEGG:E', b'7CGEFG@>', b'FEGGGGCE', b'F:GDGGGG', b'CFFGGG6@', b'CCFGGGGG', b'GGGCGGEG', b'EGFGGGGG', b'GGG@GGFG', b'GGFGGGGG', b'FEFE<FE7', b'CGGGGGGG', b'@@GGGGFG', b'EGGGGGGG', b'ECG:FGED', b'GGGGGF+8', b'@@FGCF++', b'GCGGGG+@', b'C@G:GF7C', b'++F:@E=@', b'+CFB,,>7', b'GGGGGGGG', b'FCFGGGGG', b'@6E@@<+>', b'CCF>GGEC', b'EEFC,D+8', b'FGGGGGGC', b'E:F@GG:>', b'GGGGGFGG', b'@@FEFGFE', b'CBGGGGFG', b'GGGGCFBE', b'GDFGFGGG', b'GGGGGGFG', b'@FFCGGDG', b'FECCCFBC', b'CFFGGG+>', b'DFGGGGGG', b'FGGGGGCG', b'+6A@4CFG', b'FGGDGGCE', b'CBGCFG7C', b'GGGGGGFE', b'GCDEGGFC', b'GGFGGGEG', b'GGGGGGGG', b'GGGGEGCC', b'GGGGGG:C', b'GGGGGGFG', b'GGGGGGGG', b'+6EFFGEG', b'FGGGFGCG', b'FGGGGGGG', b'CFGEGGFE', b'@6FEF<F6', b'EGGGGG=F', b'C@GGGGCF', b'GGGGGGDE', ... b'FGGGGGEG', b'CFFEGGGG', b'EGGFFG+C', b'GGFGFF:F', b'@CEF,CCF', b'CGGFGG7F', b'8FFEGGGG', b'GGGGGGCE', b'GGGGGGDG', b'FGGEGGCC', b'FEEEGG+4', b'E><@:,CF', b'GGFFFF:F', b'GGGGGGGG', b'GEGGGGGG', b'7:GCGG7F', b'CCCCGGGG', b'GEGGGGEG', b'C+FC7DEC', b'GFGEGGGG', b'GDGGGGCG', b'@+FC,C::', b'CCGCGFG+', b'FCFFFG7C', b'C@GGGG4A', b'++FF,6FF', b'+@F@<F+4', b'@:G@FFC+', b'GGGGGGEG', b'@FGGDGGG', b'+@GGF7:E', b':@GGFGFG', b'GGGGGGEG', b'FEFGGF+>', b'FEGDGG7F', b':@67?F++', b'E7ECFFFG', b'+6CF7F::', b'++C@FD+8', b'B:GFFF>F', b'C:FCFFCF', b'++,>DF=+', b'F6C@FGCC', b'C+;4GCG7', b'GFGGDFEG', b'GGFGGGFF', b'FFGGGGCG', b'+7FB4C>C', b':@GGGGFE', b'CDFFDG+@', b'++E7DF++', b'EGGGGECG', b'GGGGGGCG', b'@FEGGG4@', b'GFGGGGDG', b'GGEBGGC>', b'GGGGGGGG', b'+8GFGGCF', b'GGGGGGFG', b'C@GFGG:C', b'7:F:GGFF', b'CFGGGGGG', b'@:FFGGGG', b'F@GGFGEE', b'7CGGFGGC', b'GGGGGG@F', b'GGGGGGC@', b'GGECGGEG', b'FFFEGGC7', b'GGGGFD7B', b'CEEFGGEE', b'C@F:GF>F', b'GGE@7,FF', b'CEF7@F+=', b'GGGGGG@F', b'CCFC,DEG', b'C7G+,C++', b'+@GEGG9E', b':FDFD@C:', b'::GGFG4@', b'GFGGFF>C', b'DEFGFGCE', b'>8E7EF@=', b'7FGEGGCF', b'C7FFFG:>', b'GEC6GG++', b'EF@FGGEE', b'EDGCFGCC', b'GGGGGG@F', b'GGGGFGGG', b':7ECGGFC', b'FEGGGG>F', b'F@GG7,=F', b':FE@FFFG', b':+GCDD:C', b':CA+FG8@', b'7CGFCFF+', b'6@8C7C@B'], dtype='|S8') - sequence_coordinates(molecule, dimension)int646849 2670 9716 ... 11853 24880
array([[ 6849, 2670], [ 9716, 2565], [10664, 3332], ..., [14801, 23633], [14797, 24893], [11853, 24880]], dtype=int64) - selection_Cy5_active_start(molecule)boolFalse False True ... True False
array([False, False, True, True, True, False, False, False, False, False, False, False, True, True, False, True, False, False, True, False, False, False, True, True, False, False, False, False, False, True, True, True, False, False, False, False, True, False, True, True, True, False, True, False, True, True, True, True, True, False, False, False, False, True, False, False, False, True, True, False, True, True, True, True, True, True, False, True, False, True, False, True, True, True, True, False, True, False, True, False, True, False, True, True, False, True, False, False, False, False, True, False, False, False, False, True, True, False, True, False, True, True, True, False, True, True, True, True, True, False, True, True, True, False, True, True, False, False, False, False, True, True, False, False, True, True, True, False, True, True, False, True, True, True, True, True, False, True, True, True, False, False, True, True, True, True, False, False, False, True, True, True, True, True, False, False, False, False, True, True, False, False, False, True, True, True, True, False, True, True, False, False, False, False, False, False, True, True, True, False, ... False, True, True, True, False, False, True, False, True, False, False, True, False, False, True, False, True, True, True, True, True, False, False, False, False, True, True, True, False, False, False, False, True, True, True, False, False, True, True, True, True, True, False, False, False, False, False, False, True, True, False, True, True, False, True, True, False, False, True, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, True, True, False, True, True, False, True, True, False, False, True, False, False, False, False, False, True, False, True, True, True, False, False, False, True, True, False, True, True, True, True, True, False, True, False, False, True, False, False, False, False, True, True, True, True, False, False, True, True, True, False, False, False, True, False, True, True, False, False, False, True, True, False, True, True, True, True, False, False, True, True, True, False, True, True, True, False, False, False, False, False, True, False, True, False, False, True, True, True, True, False, False, True, True, True, True, False]) - selection_Cy5_active_end(molecule)boolFalse False True ... True False
array([False, False, True, True, True, False, False, False, False, False, False, False, True, True, False, True, False, False, True, False, False, False, True, False, False, False, False, False, False, False, True, False, False, False, False, False, True, False, True, True, True, False, True, False, True, True, True, True, True, False, False, False, False, True, False, False, False, True, True, False, False, True, False, True, True, True, False, True, False, True, False, True, True, True, True, False, True, False, True, False, True, False, True, True, False, False, False, False, False, False, True, False, False, False, False, True, True, False, True, False, False, True, True, False, True, False, True, True, False, False, True, True, True, False, True, True, False, False, False, False, True, False, False, False, True, True, True, False, True, False, False, True, True, True, True, True, False, False, True, True, False, False, True, True, True, True, False, False, False, True, True, True, True, True, False, False, False, True, False, True, False, False, False, True, True, True, True, False, True, True, False, False, False, False, False, False, True, True, True, False, ... False, True, True, True, False, False, False, False, True, False, False, False, False, False, False, False, True, True, True, True, False, False, False, False, False, True, True, True, False, False, False, False, False, True, True, False, False, True, True, False, True, True, False, False, False, False, False, False, True, True, False, True, True, False, True, True, False, False, True, True, False, False, False, False, False, True, False, True, False, False, False, True, False, True, True, True, False, True, True, True, False, True, False, True, True, False, True, False, False, False, True, False, False, False, False, False, False, False, False, True, False, False, False, False, True, True, False, True, True, True, False, True, False, False, False, False, True, False, False, False, False, False, False, True, True, False, False, True, True, True, False, False, False, False, False, True, True, False, False, False, True, True, False, True, False, True, True, False, False, True, True, True, False, True, True, True, False, False, False, False, False, True, False, True, False, False, True, True, True, True, False, False, True, True, True, True, False]) - selection_intensity_total(molecule)boolTrue True True ... True False True
array([ True, True, True, False, True, True, True, True, False, True, True, True, True, True, False, False, False, True, True, True, True, True, True, True, True, False, False, True, True, True, True, False, True, False, True, False, True, True, True, False, False, True, True, False, True, False, True, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, False, True, True, False, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, False, True, False, False, False, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, False, True, True, True, True, False, False, True, True, True, True, True, True, False, True, True, False, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, True, True, True, True, True, True, False, True, ... True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, True, False, False, False, True, True, True, True, True, True, True, False, True, True, False, False, True, True, True, True, True, False, True, True, False, True, False, False, False, False, False, False, True, True, False, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, False, True, True, True, True, True, True, True, True, False, True, False, False, True, True, True, True, False, True, True, True, True, True, False, False, True, True, True, False, True, False, True, True, True, True, True, True, True, True, True, True, True, False, True, True, True, True, True, False, False, False, True, False, True, True, True, True, True, False, True, True, True, True, True, True, True, True, True, False, True, True, True, False, True, False, True, True, True, False, True, True, False, False, True, False, True, True, True, True, False, True, True, False, True, False, True]) - classification_donor_active(molecule, frame)boolTrue True True ... True True True
array([[ True, True, True, ..., False, False, False], [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], ..., [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True]]) - classification_single_dye(molecule, frame)boolTrue True True ... True True True
array([[ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], [ True, True, True, ..., True, True, True], ..., [ True, True, True, ..., True, True, True], [False, False, False, ..., False, False, False], [ True, True, True, ..., True, True, True]])
- channelPandasIndex
PandasIndex(Int64Index([0, 1], dtype='int64', name='channel'))
- dimensionPandasIndex
PandasIndex(Index([b'x', b'y'], dtype='object', name='dimension'))
- framePandasIndex
PandasIndex(Int64Index([ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, ... 390, 391, 392, 393, 394, 395, 396, 397, 398, 399], dtype='int64', name='frame', length=400))
Here we set inactive donor frames to state -1 and frames above the total intensity treshold to state -2. These are negative states, indicating exclusion in later analysis. Anything else is set to state 0.
Applying the selections changes the classification dataarray in the dataset.
[29]:
file.apply_classifications(classification_donor_active=-1, classification_single_dye=-2)
[30]:
file.classification
[30]:
<xarray.DataArray 'classification' (molecule: 753, frame: 400)>
array([[ 0, 0, 0, ..., -1, -1, -1],
[ 0, 0, 0, ..., 0, 0, 0],
[ 0, 0, 0, ..., 0, 0, 0],
...,
[ 0, 0, 0, ..., 0, 0, 0],
[-2, -2, -2, ..., -2, -2, -2],
[ 0, 0, 0, ..., 0, 0, 0]], dtype=int8)
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
time (frame) float64 0.0 0.123 0.245 ... 48.62 48.75 48.87
* frame (frame) int32 0 1 2 3 4 5 6 ... 394 395 396 397 398 399
illumination (frame) int32 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0 0 0
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule- molecule: 753
- frame: 400
- 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0 0
array([[ 0, 0, 0, ..., -1, -1, -1], [ 0, 0, 0, ..., 0, 0, 0], [ 0, 0, 0, ..., 0, 0, 0], ..., [ 0, 0, 0, ..., 0, 0, 0], [-2, -2, -2, ..., -2, -2, -2], [ 0, 0, 0, ..., 0, 0, 0]], dtype=int8) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - time(frame)float640.0 0.123 0.245 ... 48.75 48.87
- units :
- s
array([ 0. , 0.123, 0.245, 0.368, 0.49 , 0.612, 0.735, 0.858, 0.98 , 1.102, 1.225, 1.347, 1.47 , 1.592, 1.715, 1.838, 1.96 , 2.082, 2.205, 2.327, 2.45 , 2.573, 2.694, 2.817, 2.94 , 3.062, 3.185, 3.307, 3.429, 3.552, 3.675, 3.797, 3.92 , 4.042, 4.165, 4.287, 4.41 , 4.532, 4.654, 4.777, 4.9 , 5.022, 5.145, 5.267, 5.389, 5.512, 5.635, 5.757, 5.879, 6.002, 6.124, 6.247, 6.37 , 6.491, 6.614, 6.737, 6.859, 6.982, 7.104, 7.226, 7.349, 7.472, 7.594, 7.717, 7.839, 7.961, 8.084, 8.207, 8.329, 8.451, 8.574, 8.696, 8.819, 8.942, 9.063, 9.186, 9.309, 9.431, 9.553, 9.676, 9.799, 9.921, 10.044, 10.166, 10.288, 10.411, 10.534, 10.656, 10.778, 10.901, 11.024, 11.146, 11.269, 11.391, 11.513, 11.636, 11.759, 11.881, 12.003, 12.126, 12.248, 12.371, 12.494, 12.615, 12.738, 12.861, 12.983, 13.106, 13.228, 13.35 , 13.473, 13.596, 13.718, 13.841, 13.963, 14.085, 14.208, 14.331, 14.453, 14.575, 14.698, 14.82 , 14.943, 15.066, 15.187, 15.31 , 15.433, 15.555, 15.678, 15.8 , 15.922, 16.045, 16.168, 16.291, 16.413, 16.535, 16.658, 16.78 , 16.903, 17.026, 17.147, 17.27 , 17.393, 17.515, 17.638, 17.76 , 17.882, 18.005, 18.128, 18.25 , 18.372, 18.495, 18.618, 18.74 , 18.862, 18.985, 19.107, 19.23 , 19.353, 19.474, ... 29.396, 29.518, 29.64 , 29.763, 29.886, 30.008, 30.131, 30.253, 30.375, 30.498, 30.621, 30.743, 30.865, 30.988, 31.11 , 31.233, 31.356, 31.477, 31.6 , 31.723, 31.846, 31.968, 32.09 , 32.213, 32.335, 32.458, 32.581, 32.703, 32.825, 32.948, 33.07 , 33.193, 33.316, 33.437, 33.56 , 33.683, 33.805, 33.928, 34.05 , 34.172, 34.295, 34.418, 34.54 , 34.662, 34.785, 34.907, 35.03 , 35.153, 35.274, 35.397, 35.52 , 35.643, 35.765, 35.887, 36.01 , 36.132, 36.255, 36.378, 36.499, 36.622, 36.745, 36.867, 36.99 , 37.112, 37.234, 37.357, 37.48 , 37.602, 37.725, 37.847, 37.969, 38.092, 38.215, 38.337, 38.459, 38.582, 38.704, 38.827, 38.95 , 39.071, 39.194, 39.317, 39.439, 39.562, 39.685, 39.806, 39.929, 40.052, 40.174, 40.297, 40.419, 40.541, 40.664, 40.787, 40.909, 41.031, 41.154, 41.276, 41.399, 41.522, 41.644, 41.766, 41.889, 42.012, 42.134, 42.256, 42.379, 42.501, 42.624, 42.747, 42.868, 42.991, 43.114, 43.236, 43.359, 43.482, 43.603, 43.726, 43.849, 43.971, 44.094, 44.216, 44.338, 44.461, 44.584, 44.706, 44.828, 44.951, 45.073, 45.196, 45.319, 45.44 , 45.563, 45.686, 45.809, 45.931, 46.053, 46.176, 46.298, 46.421, 46.544, 46.665, 46.788, 46.911, 47.033, 47.156, 47.278, 47.4 , 47.523, 47.646, 47.768, 47.89 , 48.013, 48.135, 48.258, 48.381, 48.502, 48.625, 48.748, 48.87 ]) - frame(frame)int320 1 2 3 4 5 ... 395 396 397 398 399
array([ 0, 1, 2, ..., 397, 398, 399])
- illumination(frame)int320 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0
array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]) - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
- framePandasIndex
PandasIndex(Int64Index([ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, ... 390, 391, 392, 393, 394, 395, 396, 397, 398, 399], dtype='int64', name='frame', length=400))
Hidden Markov modelling#
In the classify_hmm method a one-state and two-state hidden Markov model are applied to each trace and one of the two models is automatically chosen based on the Bayesian information criterion. Here, only the regions with state 0 of selected traces are used.
[31]:
file.classify_hmm('FRET')
7%|█████▏ | 49/753 [00:00<00:01, 426.89it/s]
Serial processing
100%|███████████████████████████████████████████████████████████████████████████████| 753/753 [00:03<00:00, 205.62it/s]
[32]:
file.classification_hmm
[32]:
<xarray.DataArray 'classification_hmm' (molecule: 753, frame: 400)>
array([[-1, -1, -1, ..., -1, -1, -1],
[-1, -1, -1, ..., -1, -1, -1],
[ 1, 1, 1, ..., 1, 1, 1],
...,
[ 0, 0, 0, ..., 0, 0, 0],
[-1, -1, -1, ..., -1, -1, -1],
[-1, -1, -1, ..., -1, -1, -1]], dtype=int8)
Coordinates:
molecule_in_file (molecule) int32 999 719 141 70 1113 ... 508 848 1340 352
file (molecule) |S58 b'Single-molecule data - green laser - ...
time (frame) float64 0.0 0.123 0.245 ... 48.62 48.75 48.87
* frame (frame) int32 0 1 2 3 4 5 6 ... 394 395 396 397 398 399
illumination (frame) int32 0 0 0 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0 0 0
sequence_in_file (molecule) int32 386 412 949 970 1532 ... 131 160 758 670
Dimensions without coordinates: molecule- molecule: 753
- frame: 400
- -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 ... -1 -1 -1 -1 -1 -1 -1 -1 -1 -1 -1
array([[-1, -1, -1, ..., -1, -1, -1], [-1, -1, -1, ..., -1, -1, -1], [ 1, 1, 1, ..., 1, 1, 1], ..., [ 0, 0, 0, ..., 0, 0, 0], [-1, -1, -1, ..., -1, -1, -1], [-1, -1, -1, ..., -1, -1, -1]], dtype=int8) - molecule_in_file(molecule)int32999 719 141 70 ... 508 848 1340 352
array([ 999, 719, 141, 70, 1113, 144, 251, 862, 943, 1071, 174, 736, 760, 1108, 1224, 823, 937, 262, 793, 258, 543, 770, 710, 228, 221, 394, 588, 626, 279, 910, 145, 274, 503, 512, 10, 212, 1297, 1493, 186, 178, 1483, 135, 533, 855, 1629, 1466, 1547, 1607, 55, 102, 568, 374, 331, 834, 1167, 44, 209, 726, 728, 163, 528, 5, 176, 496, 229, 836, 470, 896, 1133, 1464, 593, 635, 704, 1546, 131, 492, 620, 939, 1534, 436, 959, 587, 494, 513, 608, 374, 253, 120, 484, 552, 402, 299, 579, 698, 512, 155, 396, 329, 84, 343, 585, 485, 585, 1084, 276, 583, 1003, 1326, 861, 617, 819, 963, 355, 599, 959, 964, 188, 396, 768, 364, 631, 27, 366, 982, 1232, 255, 1483, 811, 121, 664, 435, 527, 151, 93, 302, 667, 607, 617, 662, 936, 226, 63, 123, 1183, 239, 421, 821, 579, 1312, 1377, 1257, 1368, 1464, 1488, 321, 466, 992, 691, 1109, 1120, 339, 1336, 631, 73, 208, 579, 587, 448, 2, 513, 431, 672, 1121, 603, 804, 480, 922, 1435, 787, 1117, 1432, 1514, 770, 980, 205, 881, 912, 623, 185, 601, 408, 444, 328, 1103, 358, 756, 1115, 219, 821, 1073, 422, 139, 382, 296, 738, 120, 297, 551, 834, 1181, 90, 294, 1204, 965, 1444, 577, 958, 1014, 1104, 511, ... 873, 195, 1188, 1055, 527, 739, 1267, 729, 1205, 1369, 1417, 480, 495, 504, 577, 876, 1032, 1424, 734, 908, 1085, 82, 393, 460, 808, 215, 776, 139, 496, 96, 4, 56, 328, 97, 782, 156, 369, 22, 840, 59, 924, 837, 171, 651, 485, 1106, 424, 1327, 1377, 12, 187, 233, 1184, 1636, 224, 880, 1259, 1372, 925, 842, 1356, 8, 563, 1103, 1280, 404, 299, 383, 649, 1173, 413, 514, 543, 149, 748, 965, 918, 522, 748, 256, 1177, 857, 782, 1074, 1238, 1334, 408, 908, 966, 1306, 92, 482, 32, 754, 171, 725, 255, 408, 459, 514, 128, 325, 214, 121, 166, 626, 734, 8, 374, 631, 1323, 1309, 1318, 84, 287, 464, 373, 1004, 1738, 98, 871, 678, 833, 611, 694, 908, 1675, 15, 616, 1144, 1285, 266, 655, 801, 986, 348, 1287, 710, 901, 713, 155, 495, 383, 1079, 418, 779, 1112, 223, 1296, 70, 101, 56, 815, 1088, 371, 145, 224, 189, 212, 617, 434, 579, 647, 1009, 436, 959, 1004, 75, 190, 608, 595, 469, 498, 1111, 1234, 709, 1063, 1337, 171, 1234, 1529, 126, 523, 810, 1598, 1201, 529, 603, 896, 63, 656, 841, 213, 68, 104, 645, 104, 1408, 1137, 1162, 691, 1013, 566, 320, 648, 296, 584, 513, 829, 1145, 508, 848, 1340, 352]) - file(molecule)|S58b'Single-molecule data - green l...
array([b'Single-molecule data - green laser - 001-100\\TIRF 561 0070', b'Single-molecule data - green laser - 001-100\\TIRF 561 0073', b'Single-molecule data - green laser - 001-100\\TIRF 561 0074', b'Single-molecule data - green laser - 001-100\\TIRF 561 0077', b'Single-molecule data - green laser - 001-100\\TIRF 561 0078', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0079', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0080', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0081', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0084', b'Single-molecule data - green laser - 001-100\\TIRF 561 0085', ... b'Single-molecule data - green laser - 801-896\\TIRF 561 0843', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0847', b'Single-molecule data - green laser - 801-896\\TIRF 561 0849', b'Single-molecule data - green laser - 801-896\\TIRF 561 0850', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0851', b'Single-molecule data - green laser - 801-896\\TIRF 561 0852', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0853', b'Single-molecule data - green laser - 801-896\\TIRF 561 0854', b'Single-molecule data - green laser - 801-896\\TIRF 561 0855', b'Single-molecule data - green laser - 801-896\\TIRF 561 0856', b'Single-molecule data - green laser - 801-896\\TIRF 561 0857', b'Single-molecule data - green laser - 801-896\\TIRF 561 0881', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0882', b'Single-molecule data - green laser - 801-896\\TIRF 561 0886'], dtype='|S58') - time(frame)float640.0 0.123 0.245 ... 48.75 48.87
- units :
- s
array([ 0. , 0.123, 0.245, 0.368, 0.49 , 0.612, 0.735, 0.858, 0.98 , 1.102, 1.225, 1.347, 1.47 , 1.592, 1.715, 1.838, 1.96 , 2.082, 2.205, 2.327, 2.45 , 2.573, 2.694, 2.817, 2.94 , 3.062, 3.185, 3.307, 3.429, 3.552, 3.675, 3.797, 3.92 , 4.042, 4.165, 4.287, 4.41 , 4.532, 4.654, 4.777, 4.9 , 5.022, 5.145, 5.267, 5.389, 5.512, 5.635, 5.757, 5.879, 6.002, 6.124, 6.247, 6.37 , 6.491, 6.614, 6.737, 6.859, 6.982, 7.104, 7.226, 7.349, 7.472, 7.594, 7.717, 7.839, 7.961, 8.084, 8.207, 8.329, 8.451, 8.574, 8.696, 8.819, 8.942, 9.063, 9.186, 9.309, 9.431, 9.553, 9.676, 9.799, 9.921, 10.044, 10.166, 10.288, 10.411, 10.534, 10.656, 10.778, 10.901, 11.024, 11.146, 11.269, 11.391, 11.513, 11.636, 11.759, 11.881, 12.003, 12.126, 12.248, 12.371, 12.494, 12.615, 12.738, 12.861, 12.983, 13.106, 13.228, 13.35 , 13.473, 13.596, 13.718, 13.841, 13.963, 14.085, 14.208, 14.331, 14.453, 14.575, 14.698, 14.82 , 14.943, 15.066, 15.187, 15.31 , 15.433, 15.555, 15.678, 15.8 , 15.922, 16.045, 16.168, 16.291, 16.413, 16.535, 16.658, 16.78 , 16.903, 17.026, 17.147, 17.27 , 17.393, 17.515, 17.638, 17.76 , 17.882, 18.005, 18.128, 18.25 , 18.372, 18.495, 18.618, 18.74 , 18.862, 18.985, 19.107, 19.23 , 19.353, 19.474, ... 29.396, 29.518, 29.64 , 29.763, 29.886, 30.008, 30.131, 30.253, 30.375, 30.498, 30.621, 30.743, 30.865, 30.988, 31.11 , 31.233, 31.356, 31.477, 31.6 , 31.723, 31.846, 31.968, 32.09 , 32.213, 32.335, 32.458, 32.581, 32.703, 32.825, 32.948, 33.07 , 33.193, 33.316, 33.437, 33.56 , 33.683, 33.805, 33.928, 34.05 , 34.172, 34.295, 34.418, 34.54 , 34.662, 34.785, 34.907, 35.03 , 35.153, 35.274, 35.397, 35.52 , 35.643, 35.765, 35.887, 36.01 , 36.132, 36.255, 36.378, 36.499, 36.622, 36.745, 36.867, 36.99 , 37.112, 37.234, 37.357, 37.48 , 37.602, 37.725, 37.847, 37.969, 38.092, 38.215, 38.337, 38.459, 38.582, 38.704, 38.827, 38.95 , 39.071, 39.194, 39.317, 39.439, 39.562, 39.685, 39.806, 39.929, 40.052, 40.174, 40.297, 40.419, 40.541, 40.664, 40.787, 40.909, 41.031, 41.154, 41.276, 41.399, 41.522, 41.644, 41.766, 41.889, 42.012, 42.134, 42.256, 42.379, 42.501, 42.624, 42.747, 42.868, 42.991, 43.114, 43.236, 43.359, 43.482, 43.603, 43.726, 43.849, 43.971, 44.094, 44.216, 44.338, 44.461, 44.584, 44.706, 44.828, 44.951, 45.073, 45.196, 45.319, 45.44 , 45.563, 45.686, 45.809, 45.931, 46.053, 46.176, 46.298, 46.421, 46.544, 46.665, 46.788, 46.911, 47.033, 47.156, 47.278, 47.4 , 47.523, 47.646, 47.768, 47.89 , 48.013, 48.135, 48.258, 48.381, 48.502, 48.625, 48.748, 48.87 ]) - frame(frame)int320 1 2 3 4 5 ... 395 396 397 398 399
array([ 0, 1, 2, ..., 397, 398, 399])
- illumination(frame)int320 0 0 0 0 0 0 0 ... 0 0 0 0 0 0 0 0
array([0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0, 0]) - sequence_in_file(molecule)int32386 412 949 970 ... 131 160 758 670
array([ 386, 412, 949, 970, 1532, 501, 710, 678, 564, 768, 1472, 899, 273, 765, 416, 1619, 1388, 951, 697, 149, 435, 859, 210, 502, 1187, 277, 950, 612, 973, 678, 1232, 779, 512, 1248, 627, 1202, 297, 688, 565, 1103, 1512, 1463, 1429, 1365, 826, 1115, 621, 1154, 164, 1079, 1273, 828, 969, 1105, 1307, 1282, 766, 985, 472, 523, 1192, 175, 1600, 312, 1576, 1686, 1103, 1157, 1888, 1217, 1666, 1583, 1054, 2054, 1577, 1035, 621, 472, 1772, 261, 266, 1587, 420, 1681, 653, 1720, 402, 1364, 729, 1511, 858, 144, 125, 337, 373, 124, 489, 140, 283, 481, 628, 1226, 565, 809, 945, 1417, 1592, 1510, 661, 591, 1630, 1819, 2030, 432, 440, 446, 2150, 455, 2125, 333, 1916, 1212, 1077, 1060, 516, 338, 1838, 1213, 181, 1406, 675, 187, 569, 193, 314, 648, 1147, 599, 497, 661, 936, 1517, 940, 1631, 1282, 784, 1724, 1787, 977, 722, 1647, 808, 2084, 2012, 347, 828, 1605, 1645, 1544, 1375, 1479, 1921, 971, 1151, 920, 1264, 1183, 858, 426, 785, 919, 1385, 180, 1459, 342, 898, 1176, 477, 1611, 1441, 1168, 1434, 1631, 797, 1867, 760, 1260, 1794, 837, 786, 421, 1831, 1800, 170, 241, 1273, 1105, 862, 214, 168, 803, 634, 371, 569, 105, 155, 1437, 143, 1481, 515, 971, 230, 1610, 1406, 416, 883, 1062, 1423, 1709, 334, ... 1528, 889, 1150, 270, 1012, 1259, 1057, 945, 1408, 905, 1653, 789, 1179, 1174, 406, 1756, 1414, 2049, 1443, 1359, 1736, 1688, 1361, 1004, 308, 343, 340, 536, 353, 938, 921, 522, 271, 120, 322, 350, 1080, 240, 610, 169, 226, 472, 988, 1219, 1004, 1082, 1142, 250, 1391, 440, 1376, 1951, 1109, 756, 498, 1244, 597, 2109, 1819, 1451, 449, 660, 646, 933, 1734, 1034, 777, 667, 529, 726, 354, 878, 973, 704, 169, 1352, 1474, 788, 1171, 294, 1777, 1450, 1568, 675, 1063, 432, 1263, 335, 1849, 918, 1590, 1661, 680, 600, 1288, 319, 315, 342, 814, 966, 100, 90, 111, 871, 557, 1198, 1153, 551, 637, 981, 1618, 853, 1798, 1613, 559, 1354, 1684, 1011, 1573, 279, 347, 746, 399, 810, 1571, 1452, 782, 1509, 794, 1025, 548, 1404, 1188, 994, 827, 1048, 1473, 1112, 721, 907, 341, 168, 884, 479, 361, 550, 671, 888, 683, 1509, 805, 974, 1842, 712, 1762, 1797, 567, 312, 1389, 549, 1724, 1021, 849, 1499, 1222, 339, 851, 488, 695, 844, 807, 164, 320, 956, 1069, 1303, 1473, 280, 1382, 363, 212, 1386, 523, 329, 1355, 344, 1124, 937, 1528, 1082, 662, 913, 546, 379, 99, 527, 948, 475, 691, 879, 924, 421, 799, 820, 140, 508, 527, 496, 430, 185, 131, 160, 758, 670])
- framePandasIndex
PandasIndex(Int64Index([ 0, 1, 2, 3, 4, 5, 6, 7, 8, 9, ... 390, 391, 392, 393, 394, 395, 396, 397, 398, 399], dtype='int64', name='frame', length=400))
classify_hmm adds additional selections to the dataset: - selection_complex_rates if the hmm fit produces complex-valued transition rates. - selection_lower_rate_limit if the hmm fit produces transition rates that are slower than can be measured with the time resolution.
So we apply these selections to the overall selection.
[33]:
# Apply additional selections produced by classify_hmm
file.apply_selections(['selection_intensity_total', 'selection_Cy5_active_start', 'selection_Cy5_active_end',
'selection_complex_rates', 'selection_lower_rate_limit'])
classify_hmm adds adds an additional classification to the dataset: classification_hmm. This is -1 in case the initial classification was negative, 0 in case of the low FRET state (or a single state) and 1 in case of the high FRET state.
We apply the classifications, using classification_donor_active and classification_hmm as before, but we add states 0 and 1 of classification_hmm. We do not use the -1 classification in classification_hmm, indicated by None, because this is already included in states -2 and -1.
[34]:
# Apply additional classification produced by classify_hmm
file.apply_classifications(classification_donor_active=-1, classification_single_dye=-2, classification_hmm=[None, 0, 1])
[35]:
file.plot_hmm_rates()
Evaluate traces#
[36]:
file.show_traces(plot_variables=['intensity_total', 'intensity', 'FRET', 'classification'],
ylims=[(0, 35000), (0, 35000), (0, 1), (-2.5,1.5)],
colours=[('k'), ('g', 'r'), ('b'), ('k')], selected=False, height=5)
%matplotlib inline
Full pipeline#
[37]:
def pipeline(file, red_intensity_cutoff, intensity_total_min, intensity_total_max):
try:
# Define selections
selection_Cy5_active_start = file.intensity_red_before.sel(channel=1, drop=True) > red_intensity_cutoff
selection_Cy5_active_start.name = 'selection_Cy5_active_start'
selection_Cy5_active_end = file.intensity_red_after.sel(channel=1, drop=True) > red_intensity_cutoff
selection_Cy5_active_end.name = 'selection_Cy5_active_end'
intensity_total_rolling = file.intensity_total.rolling(frame=5, center=True).mean().dropna('frame')
selection_intensity_total = (intensity_total_rolling < intensity_total_max).all('frame')
selection_intensity_total.name = 'selection_intensity_total'
# Save selections
file.set_variable(selection_Cy5_active_start)
file.set_variable(selection_Cy5_active_end)
file.set_variable(selection_intensity_total)
# Apply selections
file.apply_selections(['selection_intensity_total', 'selection_Cy5_active_start', 'selection_Cy5_active_end'])
# Define classifictions
classification_donor_active = xr.DataArray(medfilt((file.intensity_total > intensity_total_min).astype(int), kernel_size=(1,11)).astype(bool),
name='classification_donor_active',
dims=('molecule','frame'))
classification_single_dye = xr.DataArray(medfilt((file.intensity_total < intensity_total_max).astype(int), kernel_size=(1,11)).astype(bool),
name='classification_single_dye',
dims=('molecule','frame'))
# Save classifications
file.set_variable(classification_donor_active)
file.set_variable(classification_single_dye)
# Apply classifications
file.apply_classifications(classification_donor_active=-1, classification_single_dye=-2)
# Hidden markov modelling
file.classify_hmm('FRET')
# Apply additional selections produced by classify_hmm
file.apply_selections(['selection_intensity_total', 'selection_Cy5_active_start', 'selection_Cy5_active_end',
'selection_complex_rates', 'selection_lower_rate_limit'])
# Apply additional classification produced by classify_hmm
file.apply_classifications(classification_donor_active=-1, classification_single_dye=-2, classification_hmm=[None, 0, 1])
except:
print('Error:', file)
files_HJ137.serial.map(pipeline)(red_intensity_cutoff, intensity_total_min, intensity_total_max)
0%| | 0/3 [00:00<?, ?it/s]
Serial processing
100%|█████████████████████████████████████████████████████████████████████████████| 1006/1006 [00:08<00:00, 120.60it/s]
100%|███████████████████████████████████████████████████████████████████████████████| 753/753 [00:03<00:00, 210.48it/s]
100%|███████████████████████████████████████████████████████████████████████████████| 669/669 [00:06<00:00, 102.91it/s]
100%|████████████████████████████████████████████████████████████████████████████████████| 3/3 [00:22<00:00, 7.41s/it]
Plot results#
[38]:
files_HJ137.plot_hmm_rates()
0%| | 0/3 [00:00<?, ?it/s]
Serial processing
100%|████████████████████████████████████████████████████████████████████████████████████| 3/3 [00:00<00:00, 4.00it/s]